

Influenza viral neuraminidase primes bacterial coinfection through TGF-β-mediated expression of host cell receptors
- PMID: 25535343
- PMCID: PMC4291618
- DOI: 10.1073/pnas.1414422112
Influenza viral neuraminidase primes bacterial coinfection through TGF-β-mediated expression of host cell receptors
Abstract
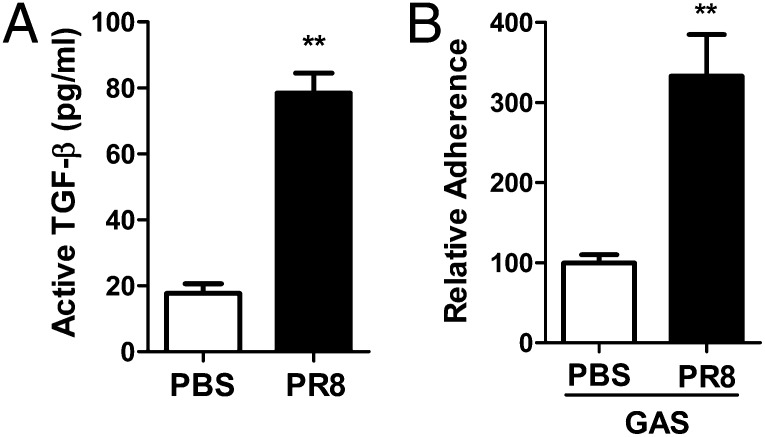
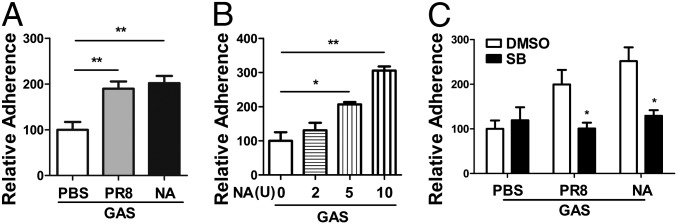
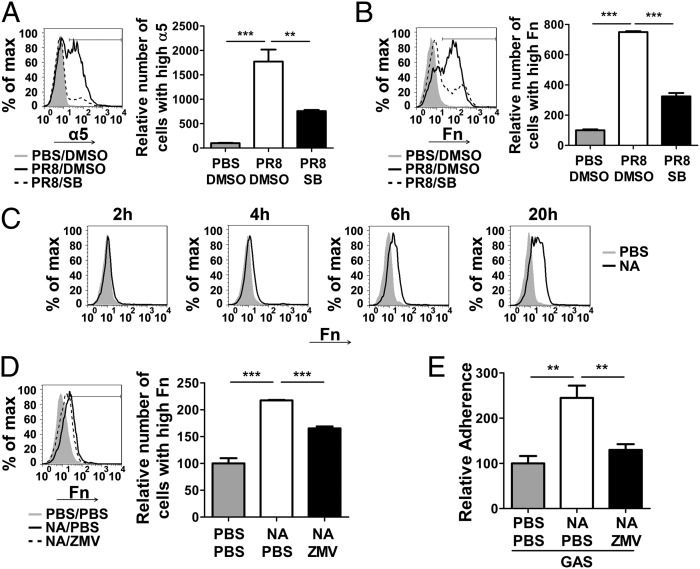
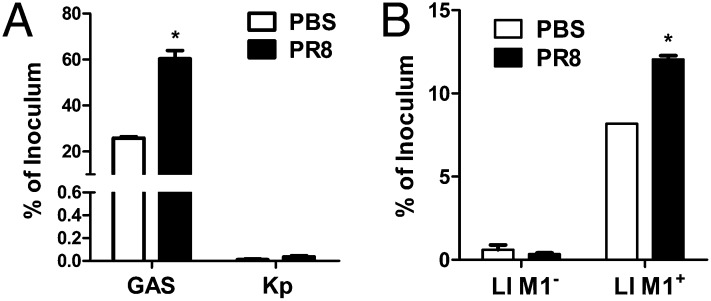
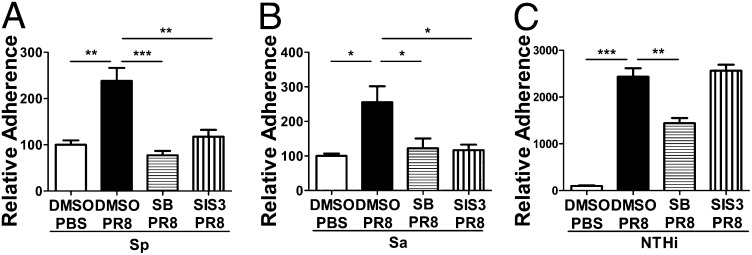
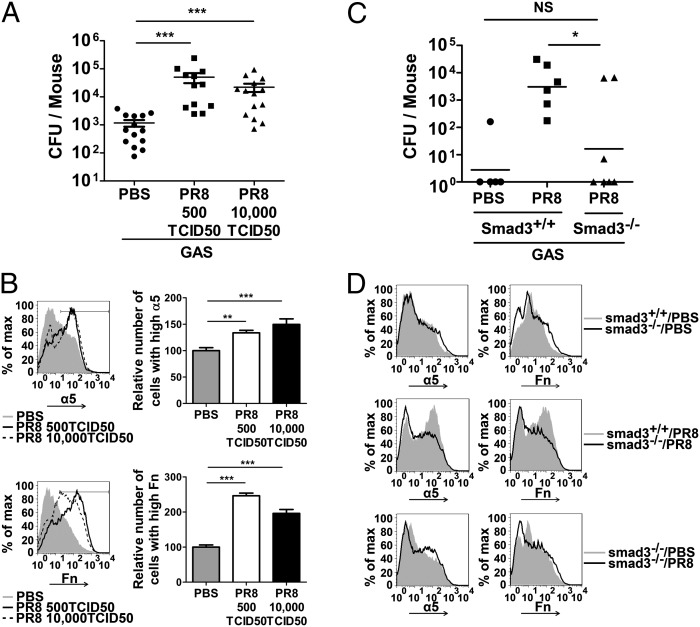
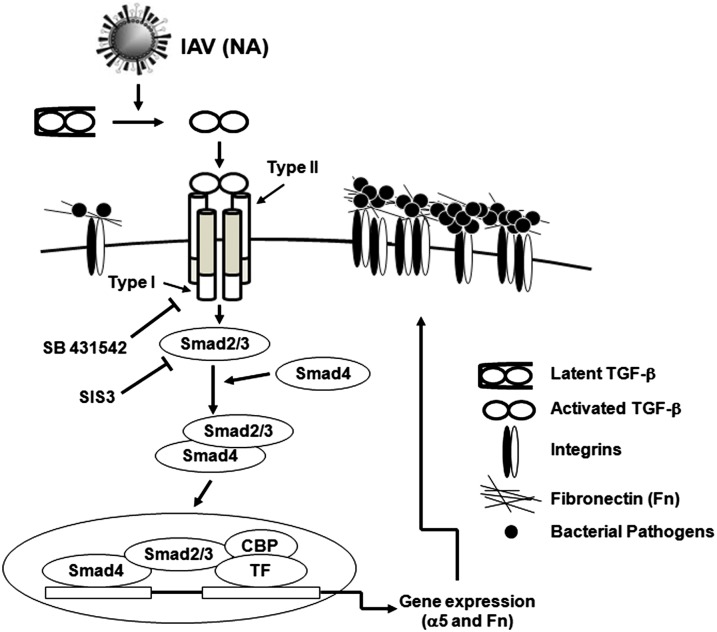
Influenza infection predisposes the host to secondary bacterial pneumonia, which is a major cause of mortality during influenza epidemics. The molecular mechanisms underlying the bacterial coinfection remain elusive. Neuraminidase (NA) of influenza A virus (IAV) enhances bacterial adherence and also activates TGF-β. Because TGF-β can up-regulate host adhesion molecules such as fibronectin and integrins for bacterial binding, we hypothesized that activated TGF-β during IAV infection contributes to secondary bacterial infection by up-regulating these host adhesion molecules. Flow cytometric analyses of a human lung epithelial cell line indicated that the expression of fibronectin and α5 integrin was up-regulated after IAV infection or treatment with recombinant NA and was reversed through the inhibition of TGF-β signaling. IAV-promoted adherence of group A Streptococcus (GAS) and other coinfective pathogens that require fibronectin for binding was prevented significantly by the inhibition of TGF-β. However, IAV did not promote the adherence of Lactococcus lactis unless this bacterium expressed the fibronectin-binding protein of GAS. Mouse experiments showed that IAV infection enhanced GAS colonization in the lungs of wild-type animals but not in the lungs of mice deficient in TGF-β signaling. Taken together, these results reveal a previously unrecognized mechanism: IAV NA enhances the expression of cellular adhesins through the activation of TGF-β, leading to increased bacterial loading in the lungs. Our results suggest that TGF-β and cellular adhesins may be potential pharmaceutical targets for the prevention of coinfection.
Keywords: TGF-beta; bacterial adherence; coinfection; fibronectin binding protein; influenza A virus.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Anonymous Centers for Disease Control and Prevention (CDC) Bacterial coinfections in lung tissue specimens from fatal cases of 2009 pandemic influenza A (H1N1) - United States, May-August 2009. MMWR Morb Mortal Wkly Rep. 2009;58(38):1071–1074. - PubMed
-
- McCullers JA. The co-pathogenesis of influenza viruses with bacteria in the lung. Nat Rev Microbiol. 2014;12(4):252–262. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
