

The dynamical nature of enzymatic catalysis
- PMID: 25539144
- PMCID: PMC4333057
- DOI: 10.1021/ar5002928
The dynamical nature of enzymatic catalysis
Abstract
CONSPECTUS: As is well-known, enzymes are proteins designed to accelerate specific life essential chemical reactions by many orders of magnitude. A folded protein is a highly dynamical entity, best described as a hierarchy or ensemble of interconverting conformations on all time scales from femtoseconds to minutes. We are just beginning to learn what role these dynamics play in the mechanism of chemical catalysis by enzymes due to extraordinary difficulties in characterizing the conformational space, that is, the energy landscape, of a folded protein. It seems clear now that their role is crucially important. Here we discuss approaches, based on vibrational spectroscopies of various sorts, that can reveal the energy landscape of an enzyme-substrate (Michaelis) complex and decipher which part of the typically very complicated landscape is relevant to catalysis. Vibrational spectroscopy is quite sensitive to small changes in bond order and bond length, with a resolution of 0.01 Å or less. It is this sensitivity that is crucial to its ability to discern bond reactivity. Using isotope edited IR approaches, we have studied in detail the role of conformational heterogeneity and dynamics in the catalysis of hydride transfer by LDH (lactate dehydrogenase). Upon the binding of substrate, the LDH·substrate system undergoes a search through conformational space to find a range of reactive conformations over the microsecond to millisecond time scale. The ligand is shuttled to the active site via first forming a weakly bound enzyme·ligand complex, probably consisting of several heterogeneous structures. This complex undergoes numerous conformational changes spread throughout the protein that shuttle the enzyme·substrate complex to a range of conformations where the substrate is tightly bound. This ensemble of conformations all have a propensity toward chemistry, but some are much more facile for carrying out chemistry than others. The search for these tightly bound states is clearly directed by the forces that the protein can bring to bear, very much akin to the folding process to form native protein in the first place. In fact, the conformational subspace of reactive conformations of the Michaelis complex can be described as a "collapse" of reactive substates compared with that found in solution, toward a much smaller and much more reactive set. These studies reveal how dynamic disorder in the protein structure can modulate the on-enzyme reactivity. It is very difficult to account for how the dynamical nature of the ground state of the Michaelis complex modulates function by transition state concepts since dynamical disorder is not a starting feature of the theory. We find that dynamical disorder may well play a larger or similar sized role in the measured Gibbs free energy of a reaction compared with the actual energy barrier involved in the chemical event. Our findings are broadly compatible with qualitative concepts of evolutionary adaptation of function such as adaptation to varying thermal environments. Our work suggests a methodology to determine the important dynamics of the Michaelis complex.
Figures
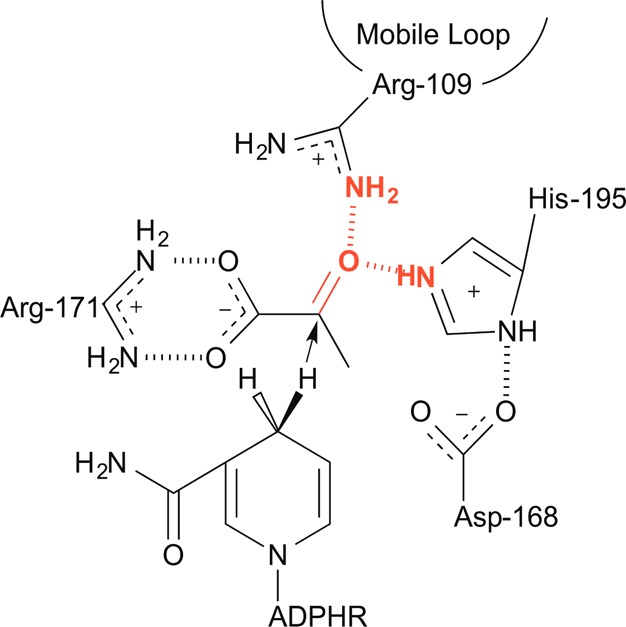
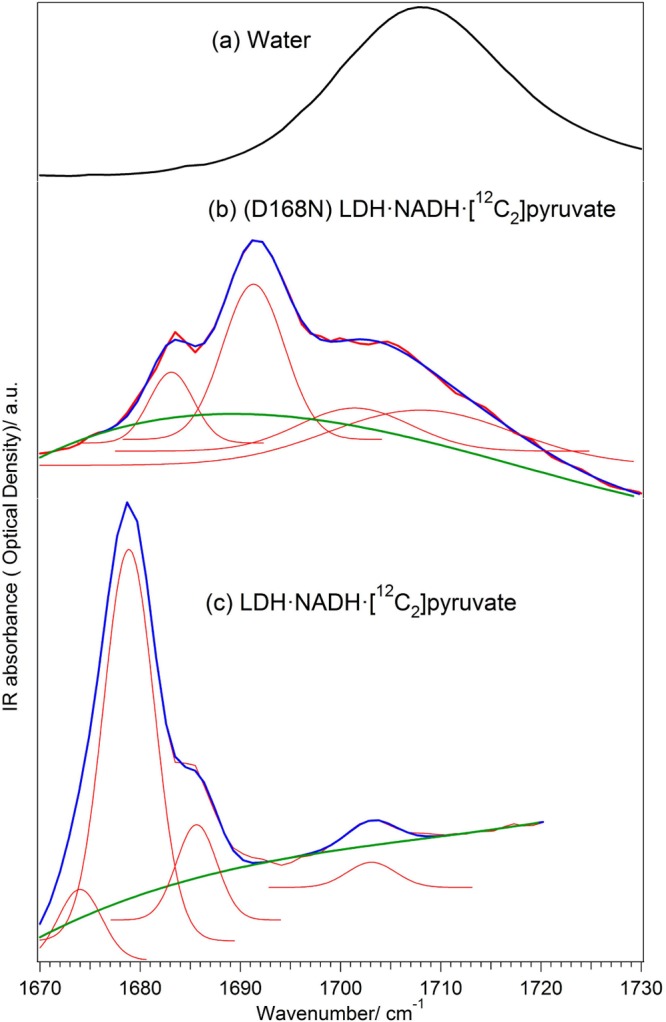
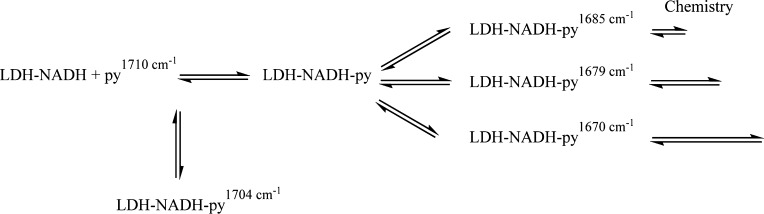
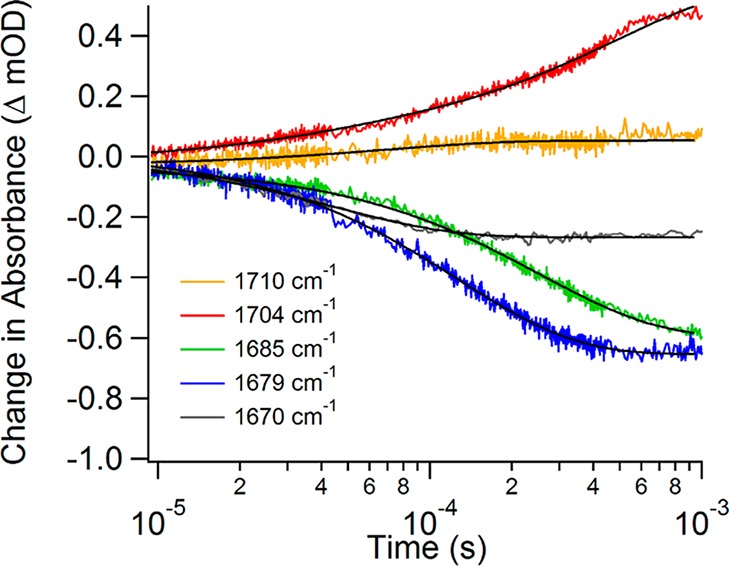
References
-
- Radzicka A.; Wolfenden R. A Proficient Enzyme. Science 1995, 267, 90–93. - PubMed
-
- Wolfenden R.; Snider M. J. The Depth of Chemical Time and Power of Enzymes as Catalysists. Acc. Chem. Res. 2001, 34, 938–945. - PubMed
-
- Arrhenius S. Ueber die Reaktionsgeschwindigkeit bei der Inversion von Rohrzucker durch Säuren. Z. Phys. Chem. 1889, 4, 226–248.
-
- Van’t Hoff J. H.Etudes de Dynamiques Chimiques; F. Muller and Co.: Amsterdam, 1884.
-
- Kramers H. A. Brownian Motion in a Field of Force and the Diffusion Model of Chemical Reactions. Physica 1940, 7, 284.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
