

Quantification of epigenetic biomarkers: an evaluation of established and emerging methods for DNA methylation analysis
- PMID: 25539843
- PMCID: PMC4523014
- DOI: 10.1186/1471-2164-15-1174
Quantification of epigenetic biomarkers: an evaluation of established and emerging methods for DNA methylation analysis
Abstract
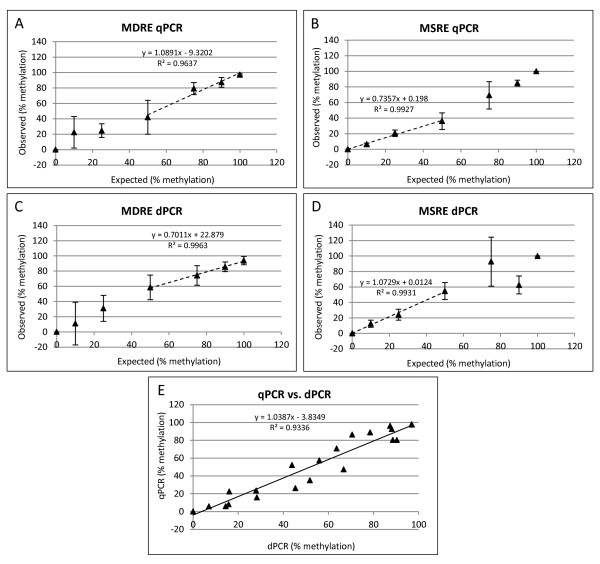
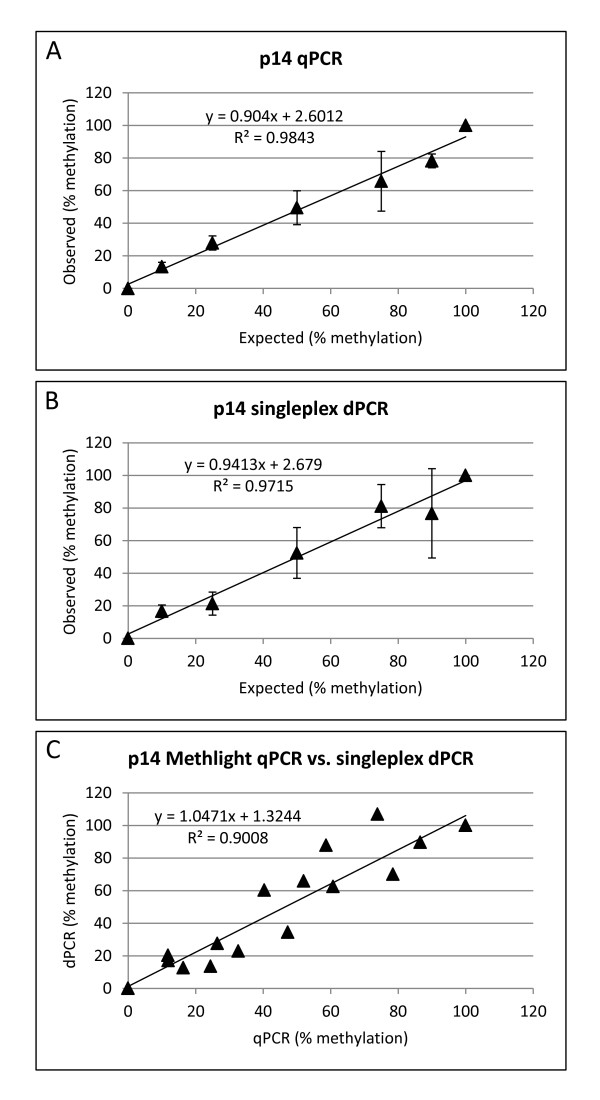
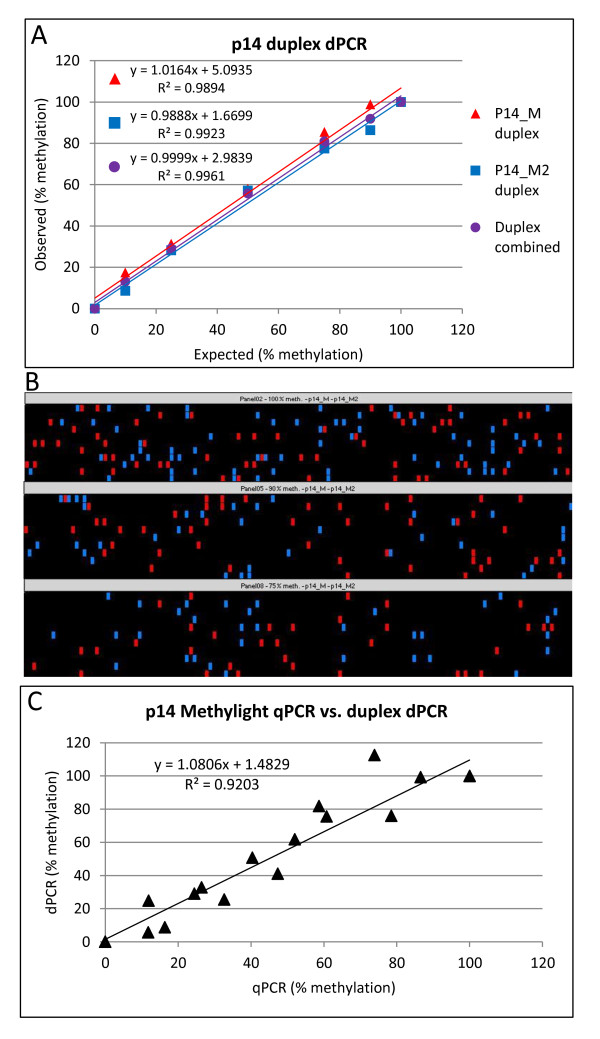
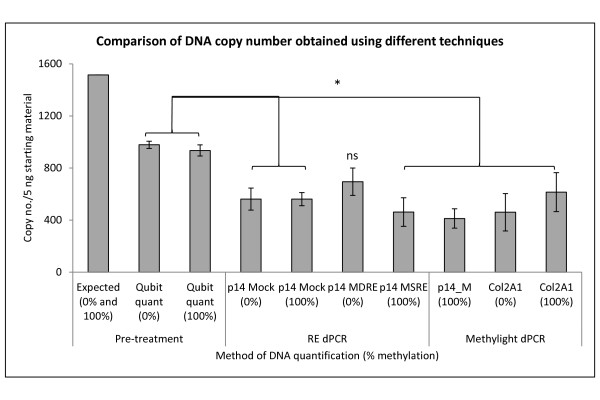
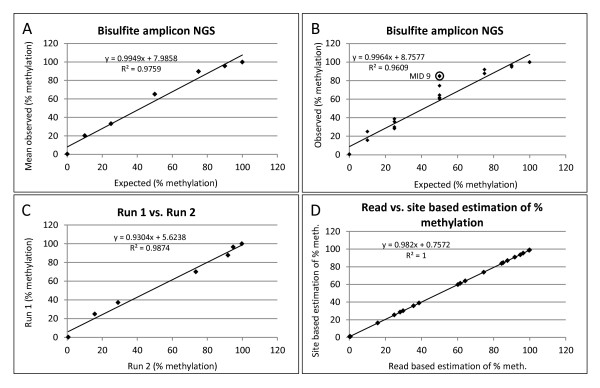
Background: DNA methylation is an important epigenetic mechanism in several human diseases, most notably cancer. The quantitative analysis of DNA methylation patterns has the potential to serve as diagnostic and prognostic biomarkers, however, there is currently a lack of consensus regarding the optimal methodologies to quantify methylation status. To address this issue we compared five analytical methods: (i) MethyLight qPCR, (ii) MethyLight digital PCR (dPCR), methylation-sensitive and -dependent restriction enzyme (MSRE/MDRE) digestion followed by (iii) qPCR or (iv) dPCR, and (v) bisulfite amplicon next generation sequencing (NGS). The techniques were evaluated for linearity, accuracy and precision.
Results: MethyLight qPCR displayed the best linearity across the range of tested samples. Observed methylation measured by MethyLight- and MSRE/MDRE-qPCR and -dPCR were not significantly different to expected values whilst bisulfite amplicon NGS analysis over-estimated methylation content. Bisulfite amplicon NGS showed good precision, whilst the lower precision of qPCR and dPCR analysis precluded discrimination of differences of < 25% in methylation status. A novel dPCR MethyLight assay is also described as a potential method for absolute quantification that simultaneously measures both sense and antisense DNA strands following bisulfite treatment.
Conclusions: Our findings comprise a comprehensive benchmark for the quantitative accuracy of key methods for methylation analysis and demonstrate their applicability to the quantification of circulating tumour DNA biomarkers by using sample concentrations that are representative of typical clinical isolates.
Figures
References
-
- Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3(6):415–428. - PubMed
-
- Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, Ji H, Potash JB, Sabunciyan S, Feinberg AP. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41(2):178–186. doi: 10.1038/ng.298. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
