

Allele Workbench: transcriptome pipeline and interactive graphics for allele-specific expression
- PMID: 25541944
- PMCID: PMC4277417
- DOI: 10.1371/journal.pone.0115740
Allele Workbench: transcriptome pipeline and interactive graphics for allele-specific expression
Abstract
Sequencing the transcriptome can answer various questions such as determining the transcripts expressed in a given species for a specific tissue or condition, evaluating differential expression, discovering variants, and evaluating allele-specific expression. Differential expression evaluates the expression differences between different strains, tissues, and conditions. Allele-specific expression evaluates expression differences between parental alleles. Both differential expression and allele-specific expression have been studied for heterosis (hybrid vigor), where the hybrid has improved performance over the parents for one or more traits. The Allele Workbench software was developed for a heterosis study that evaluated allele-specific expression for a mouse F1 hybrid using libraries from multiple tissues with biological replicates. This software has been made into a distributable package, which includes a pipeline, a Java interface to build the database, and a Java interface for query and display of the results. The required input is a reference genome, annotation file, and one or more RNA-Seq libraries with optional replicates. It evaluates allelic imbalance at the SNP and transcript level and flags transcripts with significant opposite directional allele-specific expression. The Java interface allows the user to view data from libraries, replicates, genes, transcripts, exons, and variants, including queries on allele imbalance for selected libraries. To determine the impact of allele-specific SNPs on protein folding, variants are annotated with their effect (e.g., missense), and the parental protein sequences may be exported for protein folding analysis. The Allele Workbench processing results in transcript files and read counts that can be used as input to the previously published Transcriptome Computational Workbench, which has a new algorithm for determining a trimmed set of gene ontology terms. The software with demo files is available from https://code.google.com/p/allele-workbench. Additionally, all software is ready for immediate use from an Atmosphere Virtual Machine Image available from the iPlant Collaborative (www.iplantcollaborative.org).
Conflict of interest statement
Figures
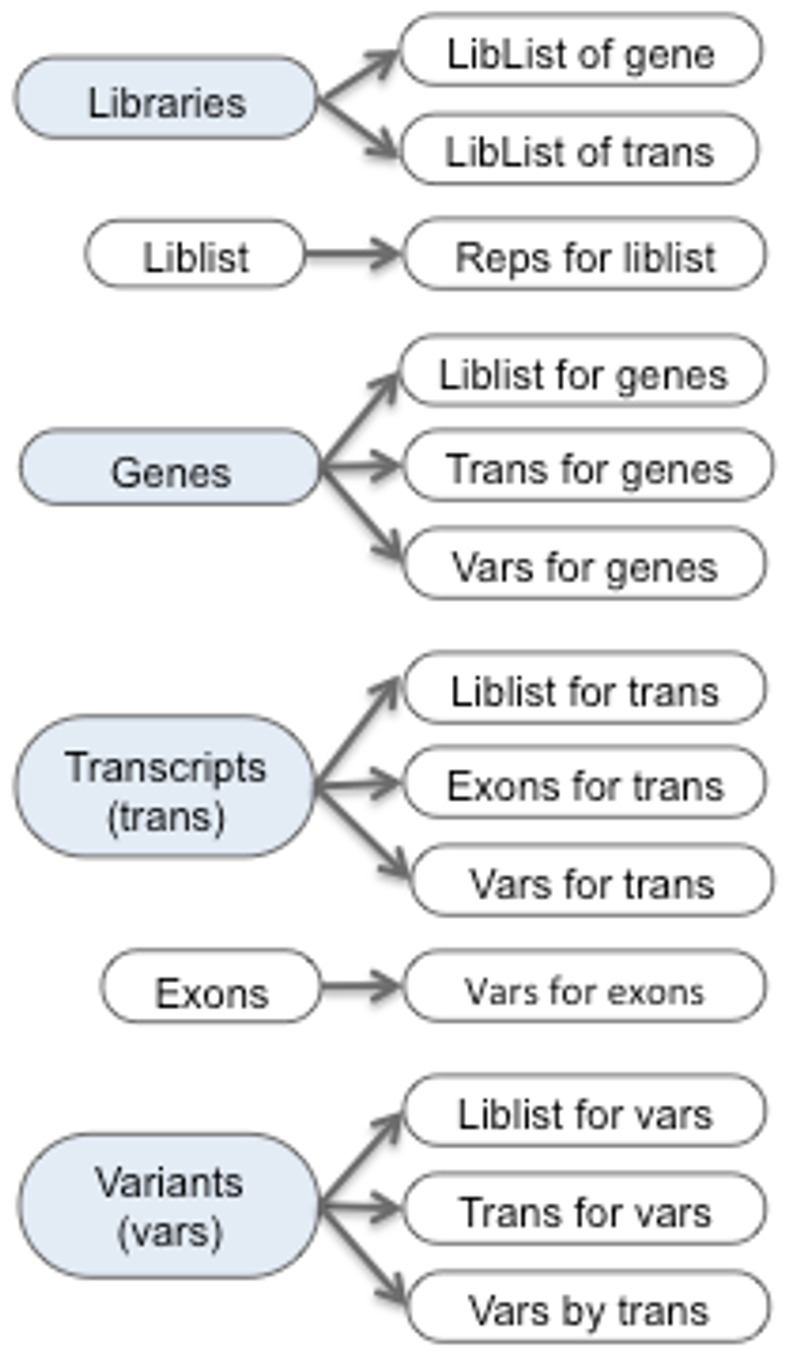
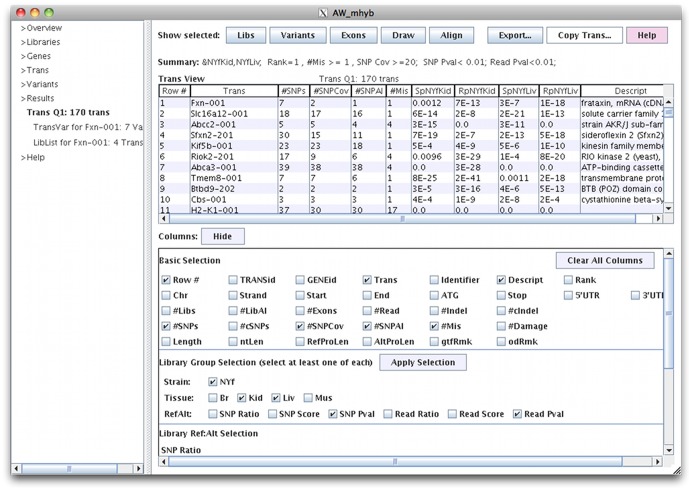
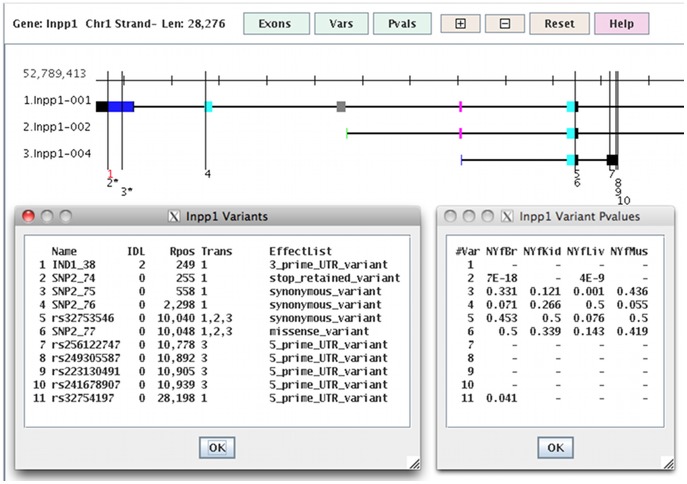
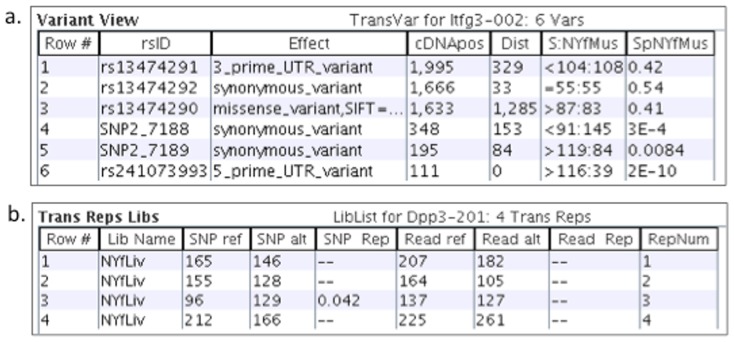
References
-
- GSNAP README (version 2013-07-16). Available: http://github.com/julian-gehring/GMAP-GSNAP/blob/master/README. Accessed 4 September 2014.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
