

Defects in mitochondrial ATP synthesis in dystrophin-deficient mdx skeletal muscles may be caused by complex I insufficiency
- PMID: 25541951
- PMCID: PMC4277356
- DOI: 10.1371/journal.pone.0115763
Defects in mitochondrial ATP synthesis in dystrophin-deficient mdx skeletal muscles may be caused by complex I insufficiency
Abstract
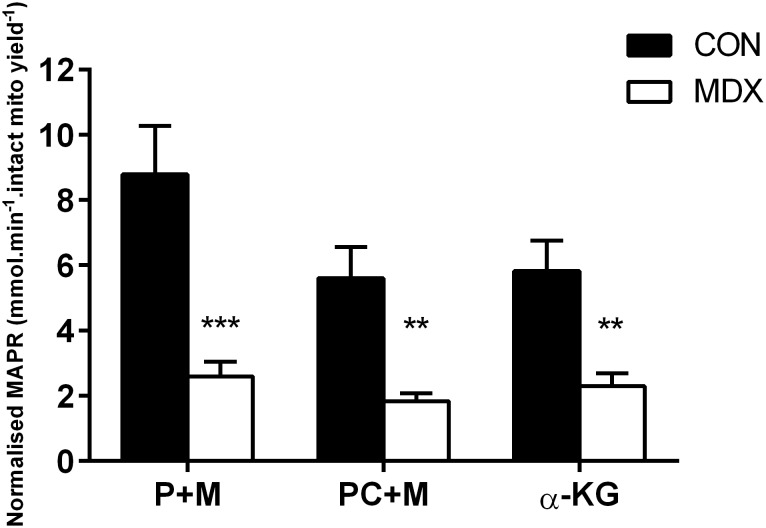
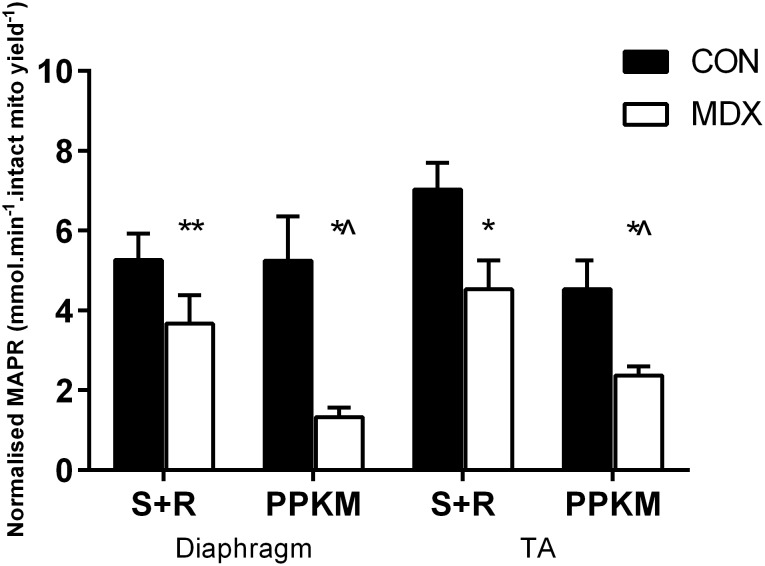
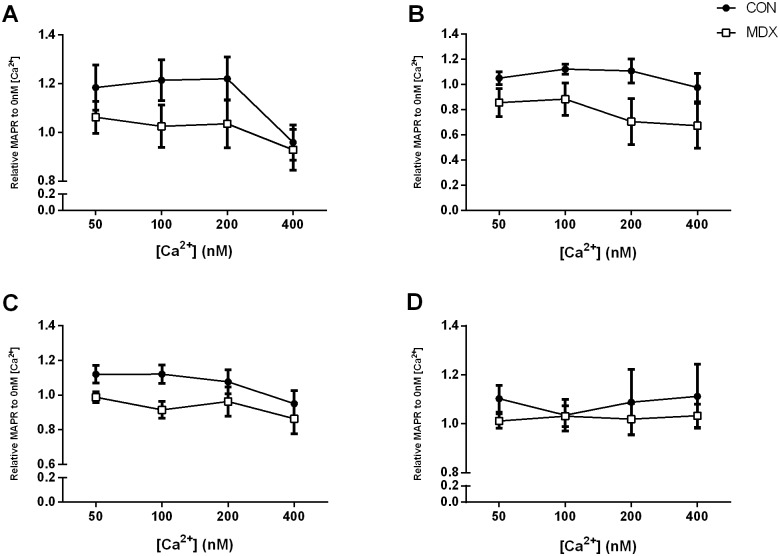
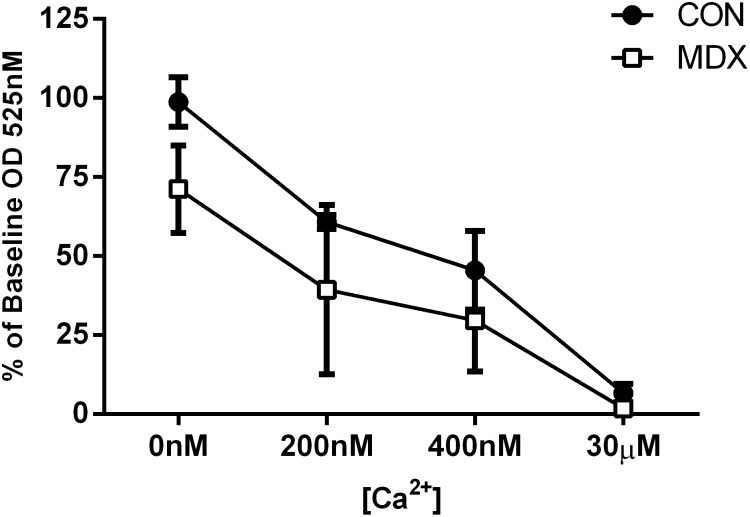
Duchenne Muscular Dystrophy is a chronic, progressive and ultimately fatal skeletal muscle wasting disease characterised by sarcolemmal fragility and intracellular Ca2+ dysregulation secondary to the absence of dystrophin. Mounting literature also suggests that the dysfunction of key energy systems within the muscle may contribute to pathological muscle wasting by reducing ATP availability to Ca2+ regulation and fibre regeneration. No study to date has biochemically quantified and contrasted mitochondrial ATP production capacity by dystrophic mitochondria isolated from their pathophysiological environment such to determine whether mitochondria are indeed capable of meeting this heightened cellular ATP demand, or examined the effects of an increasing extramitochondrial Ca2+ environment. Using isolated mitochondria from the diaphragm and tibialis anterior of 12 week-old dystrophin-deficient mdx and healthy control mice (C57BL10/ScSn) we have demonstrated severely depressed Complex I-mediated mitochondrial ATP production rate in mdx mitochondria that occurs irrespective of the macronutrient-derivative substrate combination fed into the Kreb's cycle, and, which is partially, but significantly, ameliorated by inhibition of Complex I with rotenone and stimulation of Complex II-mediated ATP-production with succinate. There was no difference in the MAPR response of mdx mitochondria to increasing extramitochondrial Ca2+ load in comparison to controls, and 400 nM extramitochondrial Ca2+ was generally shown to be inhibitory to MAPR in both groups. Our data suggests that DMD pathology is exacerbated by a Complex I deficiency, which may contribute in part to the severe reductions in ATP production previously observed in dystrophic skeletal muscle.
Conflict of interest statement
Figures
References
-
- Hoffman EP, Brown RH, Kunkel LM (1987) Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51:919–928. - PubMed
-
- Eagle M, Baudouin SV, Chandler C, Giddings DR, Bullock R, et al. (2002) Survival in Duchenne muscular dystrophy: improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscular Disorders 12:926–929. - PubMed
-
- Austin L, De Niese M, McGregor A, Arthur H, Gurusinghe A, et al. (1992) Potential oxyradical damage and energy status in individual muscle fibres from degenerating muscle diseases. Neuromuscular Disorders 2:27–33. - PubMed
-
- Cole M, Rafael J, Taylor D, Lodi R, Davies K, et al. (2002) A quantitative study of bioenergetics in skeletal muscle lacking utrophin and dystrophin. Neuromuscular Disorders 12:247–257. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
