

Natural history of β-cell adaptation and failure in type 2 diabetes
- PMID: 25542976
- PMCID: PMC4404183
- DOI: 10.1016/j.mam.2014.12.002
Natural history of β-cell adaptation and failure in type 2 diabetes
Abstract
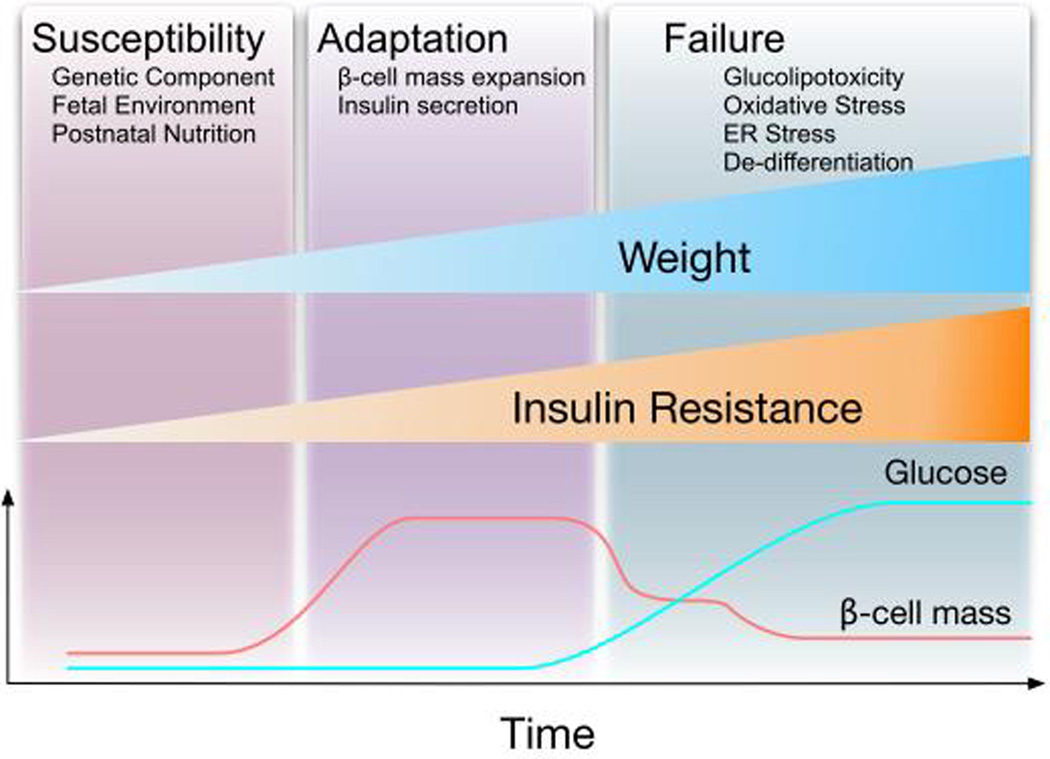
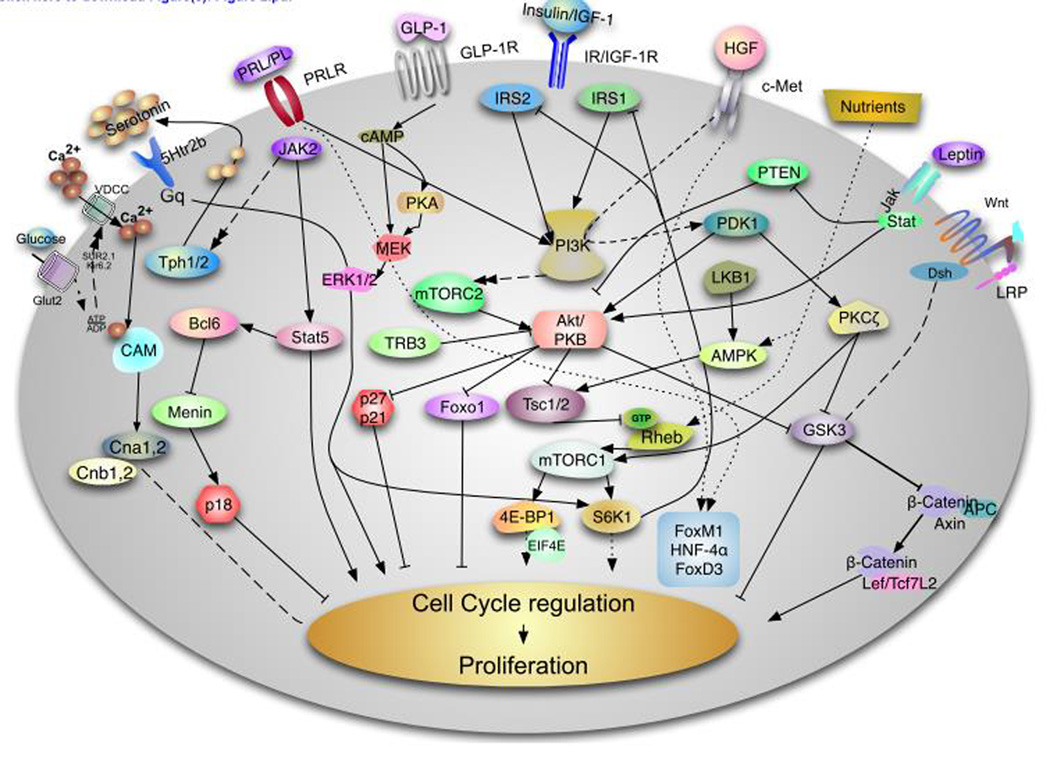
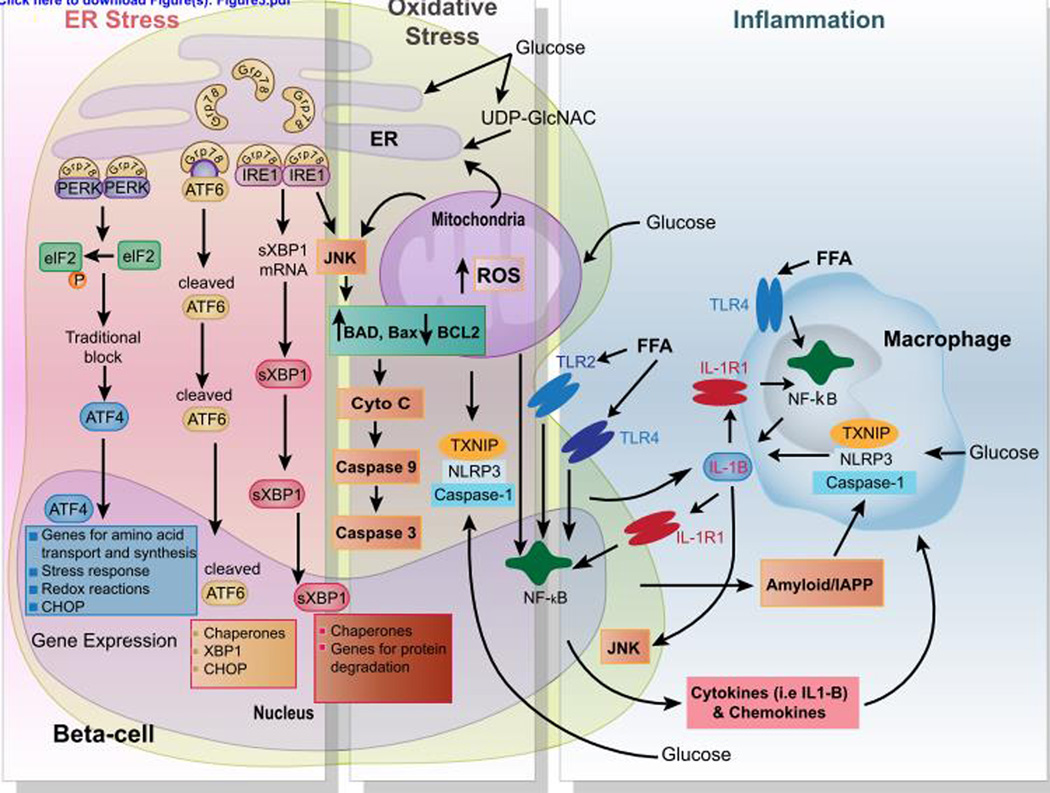
Type 2 diabetes mellitus (T2D) is a complex disease characterized by β-cell failure in the setting of insulin resistance. The current evidence suggests that genetic predisposition, and environmental factors can impair the capacity of the β-cells to respond to insulin resistance and ultimately lead to their failure. However, genetic studies have demonstrated that known variants account for less than 10% of the overall estimated T2D risk, suggesting that additional unidentified factors contribute to susceptibility of this disease. In this review, we will discuss the different stages that contribute to the development of β-cell failure in T2D. We divide the natural history of this process in three major stages: susceptibility, β-cell adaptation and β-cell failure, and provide an overview of the molecular mechanisms involved. Further research into mechanisms will reveal key modulators of β-cell failure and thus identify possible novel therapeutic targets and potential interventions to protect against β-cell failure.
Keywords: Glucolipotoxicity; Insulin resistance; Islet biology; β-cell development; β-cell failure; β-cell programming.
Published by Elsevier Ltd.
Figures
References
-
- Mokdad AH, et al. The continuing epidemics of obesity and diabetes in the United States. JAMA : the journal of the American Medical Association. 2001;286:1195–1200. - PubMed
-
- Must A, et al. The disease burden associated with overweight and obesity. JAMA : the journal of the American Medical Association. 1999;282:1523–1529. - PubMed
-
- Narayan KM, Boyle JP, Thompson TJ, Gregg EW, Williamson DF. Effect of BMI on lifetime risk for diabetes in the U.S. Diabetes care. 2007;30:1562–1566. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
