

Fanconi anemia gene editing by the CRISPR/Cas9 system
- PMID: 25545896
- PMCID: PMC4326027
- DOI: 10.1089/hum.2014.111
Fanconi anemia gene editing by the CRISPR/Cas9 system
Abstract
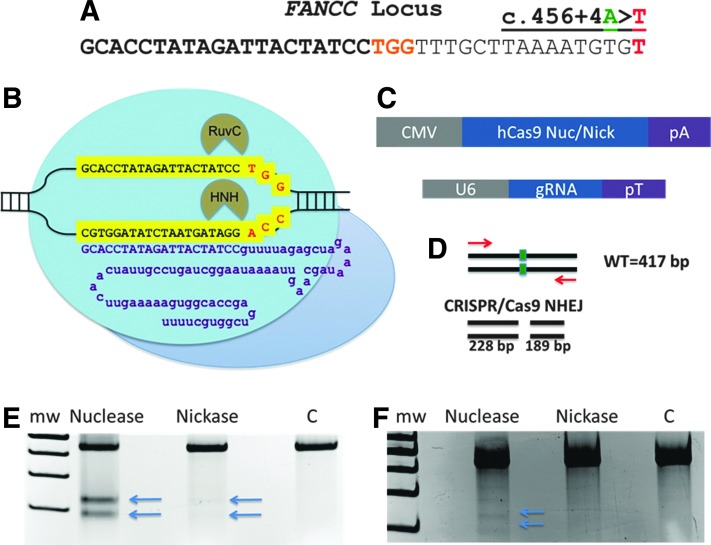
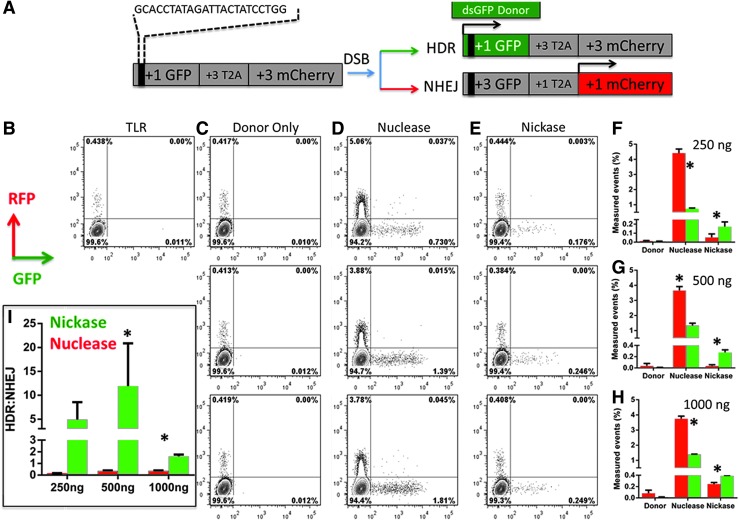
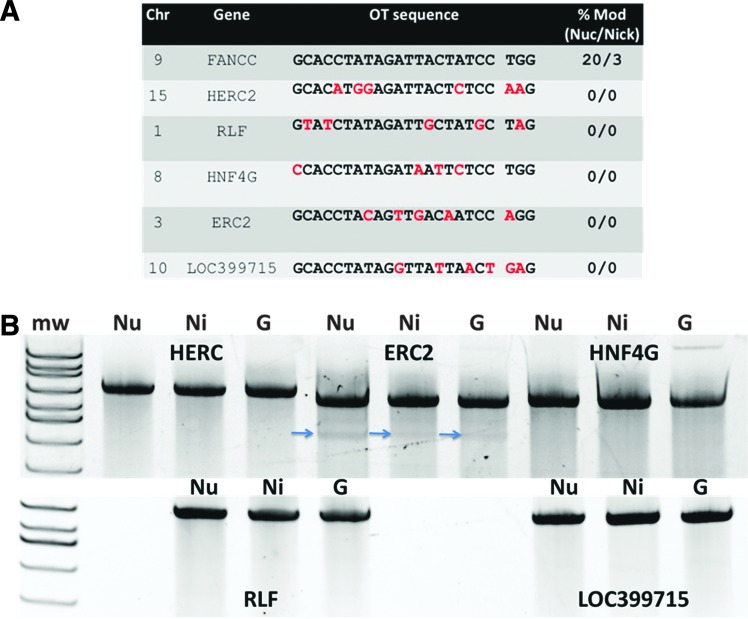
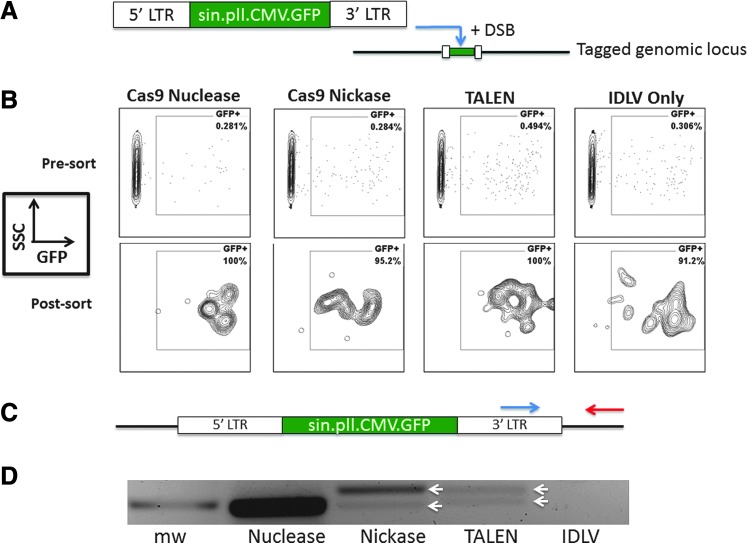
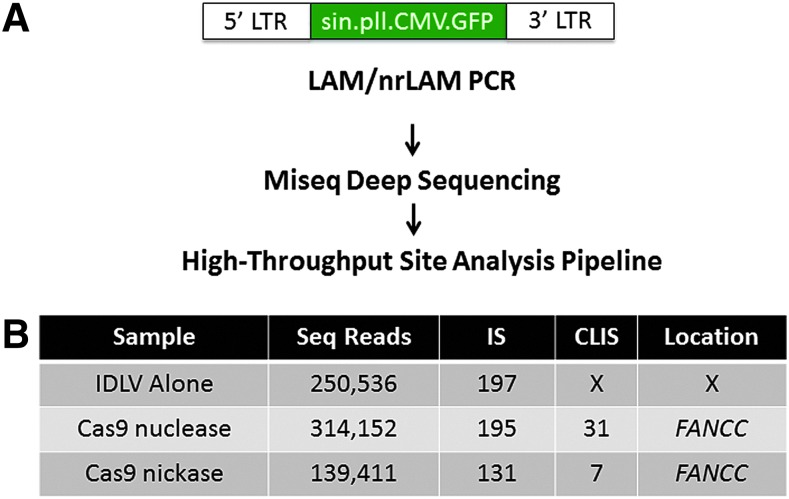
Genome engineering with designer nucleases is a rapidly progressing field, and the ability to correct human gene mutations in situ is highly desirable. We employed fibroblasts derived from a patient with Fanconi anemia as a model to test the ability of the clustered regularly interspaced short palindromic repeats/Cas9 nuclease system to mediate gene correction. We show that the Cas9 nuclease and nickase each resulted in gene correction, but the nickase, because of its ability to preferentially mediate homology-directed repair, resulted in a higher frequency of corrected clonal isolates. To assess the off-target effects, we used both a predictive software platform to identify intragenic sequences of homology as well as a genome-wide screen utilizing linear amplification-mediated PCR. We observed no off-target activity and show RNA-guided endonuclease candidate sites that do not possess low sequence complexity function in a highly specific manner. Collectively, we provide proof of principle for precision genome editing in Fanconi anemia, a DNA repair-deficient human disorder.
Figures


References
-
- Huard CC, Tremblay CS, Helsper K, et al. Fanconi anemia proteins interact with CtBP1 and modulate the expression of the Wnt antagonist Dickkopf-1. Blood 2013;121:1729–1739 - PubMed
-
- Whitney MA, Jakobs P, Kaback M, et al. The Ashkenazi Jewish Fanconi anemia mutation: incidence among patients and carrier frequency in the at-risk population. Hum Mutat 1994;3:339–341 - PubMed
-
- Whitney MA, Saito H, Jakobs PM, et al. A common mutation in the FACC gene causes Fanconi anaemia in Ashkenazi Jews. Nat Genet 1993;4:202–205 - PubMed
-
- Grompe M, D'Andrea A. Fanconi anemia and DNA repair. Hum Mol Genet 2001;10:2253–2259 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials