

DNA polymerase β deficiency leads to neurodegeneration and exacerbates Alzheimer disease phenotypes
- PMID: 25552414
- PMCID: PMC4333403
- DOI: 10.1093/nar/gku1356
DNA polymerase β deficiency leads to neurodegeneration and exacerbates Alzheimer disease phenotypes
Abstract
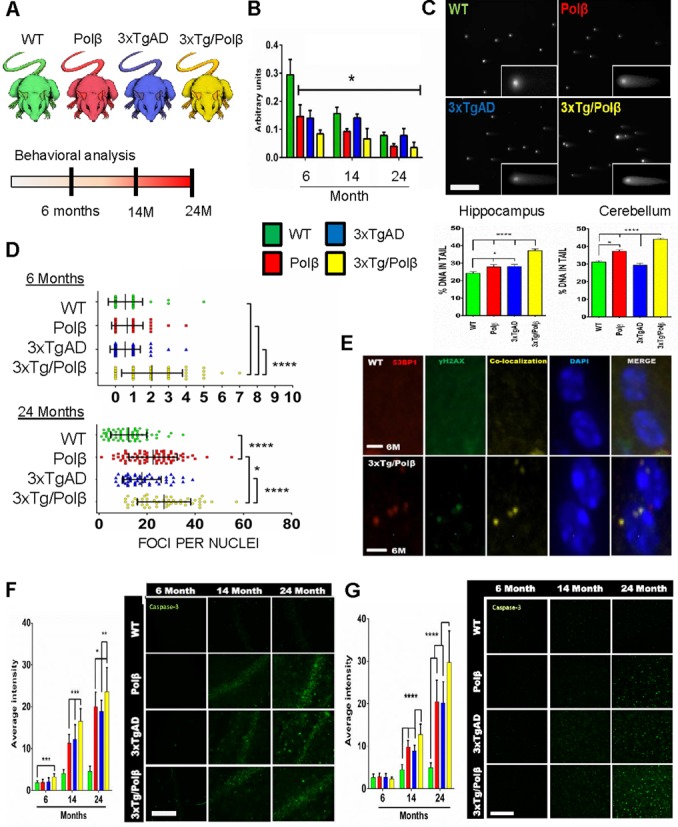
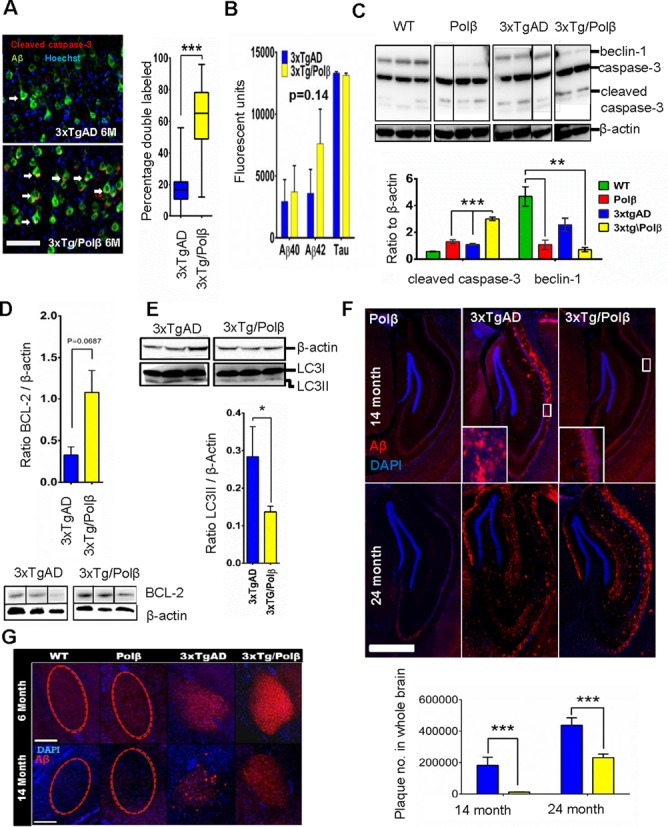
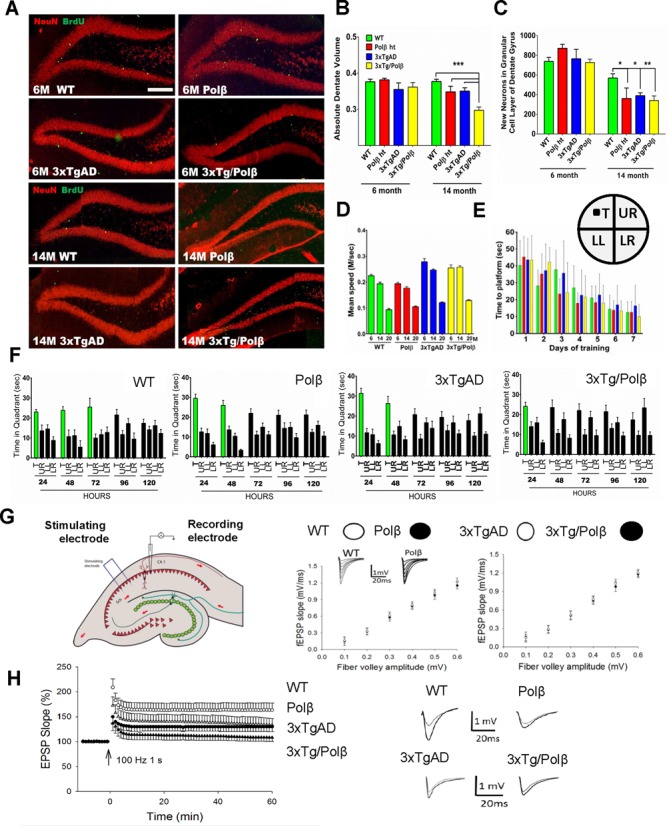
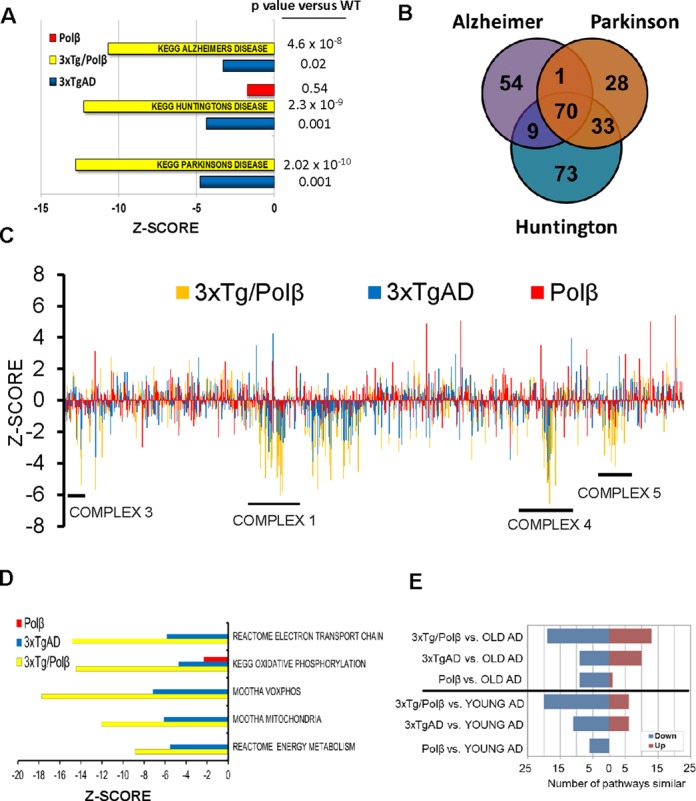
We explore the role of DNA damage processing in the progression of cognitive decline by creating a new mouse model. The new model is a cross of a common Alzheimer's disease (AD) mouse (3xTgAD), with a mouse that is heterozygous for the critical DNA base excision repair enzyme, DNA polymerase β. A reduction of this enzyme causes neurodegeneration and aggravates the AD features of the 3xTgAD mouse, inducing neuronal dysfunction, cell death and impairing memory and synaptic plasticity. Transcriptional profiling revealed remarkable similarities in gene expression alterations in brain tissue of human AD patients and 3xTg/Polβ(+/-) mice including abnormalities suggestive of impaired cellular bioenergetics. Our findings demonstrate that a modest decrement in base excision repair capacity can render the brain more vulnerable to AD-related molecular and cellular alterations.
Published by Oxford University Press on behalf of Nucleic Acids Research 2014. This work is written by US Government employees and is in the public domain in the US.
Figures
Similar articles
-
DNA polymerase β decrement triggers death of olfactory bulb cells and impairs olfaction in a mouse model of Alzheimer's disease.Aging Cell. 2017 Feb;16(1):162-172. doi: 10.1111/acel.12541. Epub 2016 Sep 30. Aging Cell. 2017. PMID: 27686631 Free PMC article.
-
NAD+ supplementation normalizes key Alzheimer's features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency.Proc Natl Acad Sci U S A. 2018 Feb 20;115(8):E1876-E1885. doi: 10.1073/pnas.1718819115. Epub 2018 Feb 5. Proc Natl Acad Sci U S A. 2018. PMID: 29432159 Free PMC article.
-
Transient OGG1, APE1, PARP1 and Polβ expression in an Alzheimer's disease mouse model.Mech Ageing Dev. 2013 Oct;134(10):467-77. doi: 10.1016/j.mad.2013.09.002. Epub 2013 Oct 11. Mech Ageing Dev. 2013. PMID: 24121118
-
Effect of Dietary Ketosis and Nicotinamide Riboside on Hippocampal Krebs Cycle Intermediates and Mitochondrial Energetics in a DNA Repair-Deficient 3xTg/POLβ+/- Alzheimer Disease Mouse Model.J Neurochem. 2025 Jan;169(1):e16295. doi: 10.1111/jnc.16295. J Neurochem. 2025. PMID: 39788884 Free PMC article.
-
Effects of CX3CR1 and Fractalkine Chemokines in Amyloid Beta Clearance and p-Tau Accumulation in Alzheimer's Disease (AD) Rodent Models: Is Fractalkine a Systemic Biomarker for AD?Curr Alzheimer Res. 2016;13(4):403-12. doi: 10.2174/1567205013666151116125714. Curr Alzheimer Res. 2016. PMID: 26567742 Review.
Cited by
-
Genome Integrity and Neurological Disease.Int J Mol Sci. 2022 Apr 8;23(8):4142. doi: 10.3390/ijms23084142. Int J Mol Sci. 2022. PMID: 35456958 Free PMC article. Review.
-
Research Progress on the Anti-Aging Potential of the Active Components of Ginseng.Nutrients. 2023 Jul 25;15(15):3286. doi: 10.3390/nu15153286. Nutrients. 2023. PMID: 37571224 Free PMC article. Review.
-
Mitochondrial base excision repair positively correlates with longevity in the liver and heart of mammals.Geroscience. 2020 Apr;42(2):653-665. doi: 10.1007/s11357-020-00158-4. Epub 2020 Jan 22. Geroscience. 2020. PMID: 31970600 Free PMC article.
-
Dysregulation of energy metabolism in Alzheimer's disease.J Neurol. 2024 Dec 2;272(1):2. doi: 10.1007/s00415-024-12800-8. J Neurol. 2024. PMID: 39621206 Free PMC article. Review.
-
Aging Neurovascular Unit and Potential Role of DNA Damage and Repair in Combating Vascular and Neurodegenerative Disorders.Front Neurosci. 2019 Aug 8;13:778. doi: 10.3389/fnins.2019.00778. eCollection 2019. Front Neurosci. 2019. PMID: 31440124 Free PMC article. Review.
References
-
- Gabbita S.P., Lovell M.A., Markesbery W.R. Increased nuclear DNA oxidation in the brain in Alzheimer's disease. J. Neurochem. 1998;71:2034–2040. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
