

The GSK-3 family as therapeutic target for myocardial diseases
- PMID: 25552693
- PMCID: PMC4283216
- DOI: 10.1161/CIRCRESAHA.116.303613
The GSK-3 family as therapeutic target for myocardial diseases
Abstract
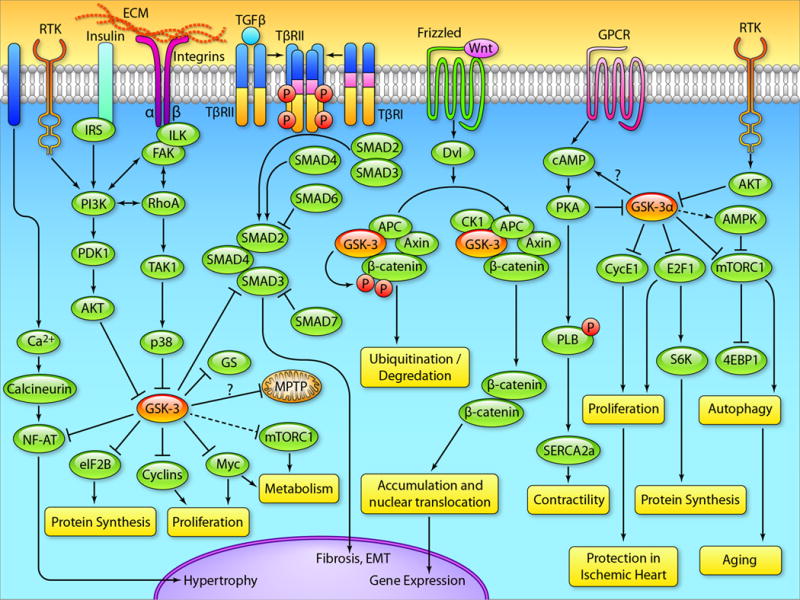
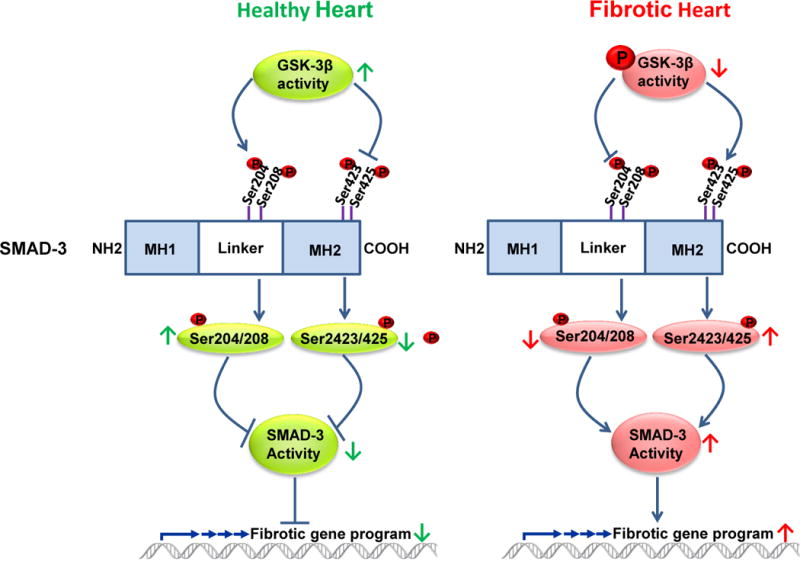
Glycogen synthase kinase-3 (GSK-3) is one of the few signaling molecules that regulate a truly astonishing number of critical intracellular signaling pathways. It has been implicated in several diseases including heart failure, bipolar disorder, diabetes mellitus, Alzheimer disease, aging, inflammation, and cancer. Furthermore, a recent clinical trial has validated the feasibility of targeting GSK-3 with small molecule inhibitors for human diseases. In the current review, we will focus on its expanding role in the heart, concentrating primarily on recent studies that have used cardiomyocyte- and fibroblast-specific conditional gene deletion in mouse models. We will highlight the role of the GSK-3 isoforms in various pathological conditions including myocardial aging, ischemic injury, myocardial fibrosis, and cardiomyocyte proliferation. We will discuss our recent findings that deletion of GSK-3α specifically in cardiomyocytes attenuates ventricular remodeling and cardiac dysfunction after myocardial infarction by limiting scar expansion and promoting cardiomyocyte proliferation. The recent emergence of GSK-3β as a regulator of myocardial fibrosis will also be discussed. We will review our recent findings that specific deletion of GSK-3β in cardiac fibroblasts leads to fibrogenesis, left ventricular dysfunction, and excessive scarring in the ischemic heart. Finally, we will examine the underlying mechanisms that drive the aberrant myocardial fibrosis in the models in which GSK-3β is specifically deleted in cardiac fibroblasts. We will summarize these recent results and offer explanations, whenever possible, and hypotheses when not. For these studies we will rely heavily on our models and those of others to reconcile some of the apparent inconsistencies in the literature.
Keywords: fibrosis; glycogen synthase kinase-3; heart failure; myocardial infarction.
© 2015 American Heart Association, Inc.
Figures
Similar articles
-
Cardiac fibroblast glycogen synthase kinase-3β regulates ventricular remodeling and dysfunction in ischemic heart.Circulation. 2014 Jul 29;130(5):419-30. doi: 10.1161/CIRCULATIONAHA.113.008364. Epub 2014 Jun 4. Circulation. 2014. PMID: 24899689 Free PMC article.
-
GSK-3 at the heart of cardiometabolic diseases: Isoform-specific targeting is critical to therapeutic benefit.Biochim Biophys Acta Mol Basis Dis. 2023 Aug;1869(6):166724. doi: 10.1016/j.bbadis.2023.166724. Epub 2023 Apr 23. Biochim Biophys Acta Mol Basis Dis. 2023. PMID: 37094727 Free PMC article. Review.
-
Crosstalk between GSK-3β-actuated molecular cascades and myocardial physiology.Heart Fail Rev. 2021 Nov;26(6):1495-1504. doi: 10.1007/s10741-020-09961-9. Heart Fail Rev. 2021. PMID: 32314086 Review.
-
Cardiac fibroblast GSK-3α aggravates ischemic cardiac injury by promoting fibrosis, inflammation, and impairing angiogenesis.Basic Res Cardiol. 2023 Sep 1;118(1):35. doi: 10.1007/s00395-023-01005-1. Basic Res Cardiol. 2023. PMID: 37656238 Free PMC article.
-
Fibroblast GSK-3α Promotes Fibrosis via RAF-MEK-ERK Pathway in the Injured Heart.Circ Res. 2022 Sep 16;131(7):620-636. doi: 10.1161/CIRCRESAHA.122.321431. Epub 2022 Sep 2. Circ Res. 2022. PMID: 36052698 Free PMC article.
Cited by
-
Genome Editing and Cardiac Regeneration.Adv Exp Med Biol. 2023;1396:37-52. doi: 10.1007/978-981-19-5642-3_3. Adv Exp Med Biol. 2023. PMID: 36454458
-
The insulin receptor family and protein kinase B (Akt) are activated in the heart by alkaline pH and α1-adrenergic receptors.Biochem J. 2021 Jun 11;478(11):2059-2079. doi: 10.1042/BCJ20210144. Biochem J. 2021. PMID: 34002209 Free PMC article.
-
Isoform- and Cell Type-Specific Roles of Glycogen Synthase Kinase 3 N-Terminal Serine Phosphorylation in Liver Ischemia Reperfusion Injury.J Immunol. 2020 Aug 15;205(4):1147-1156. doi: 10.4049/jimmunol.2000397. Epub 2020 Jul 17. J Immunol. 2020. PMID: 32680958 Free PMC article.
-
Reprogramming of cardiac phosphoproteome, proteome, and transcriptome confers resilience to chronic adenylyl cyclase-driven stress.Elife. 2024 Jan 22;12:RP88732. doi: 10.7554/eLife.88732. Elife. 2024. PMID: 38251682 Free PMC article.
-
Aberrant Neuronal Cell Cycle Re-Entry: The Pathological Confluence of Alzheimer's Disease and Brain Insulin Resistance, and Its Relation to Cancer.J Alzheimers Dis. 2019;67(1):1-11. doi: 10.3233/JAD-180874. J Alzheimers Dis. 2019. PMID: 30452418 Free PMC article. Review.
References
-
- Lochhead PA, Kinstrie R, Sibbet G, Rawjee T, Morrice N, Cleghon V. A chaperone-dependent GSK3beta transitional intermediate mediates activation-loop autophosphorylation. Mol Cell. 2006;24:627–33. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
