

Early etiology of Alzheimer's disease: tipping the balance toward autophagy or endosomal dysfunction?
- PMID: 25556159
- PMCID: PMC4331606
- DOI: 10.1007/s00401-014-1379-7
Early etiology of Alzheimer's disease: tipping the balance toward autophagy or endosomal dysfunction?
Abstract
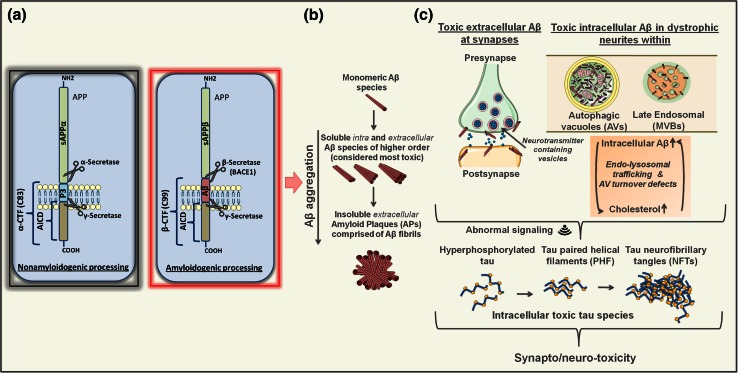
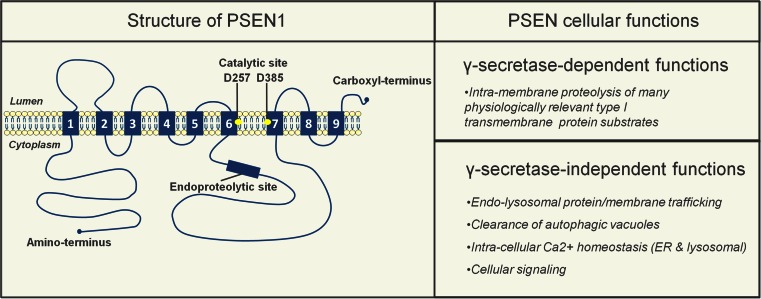
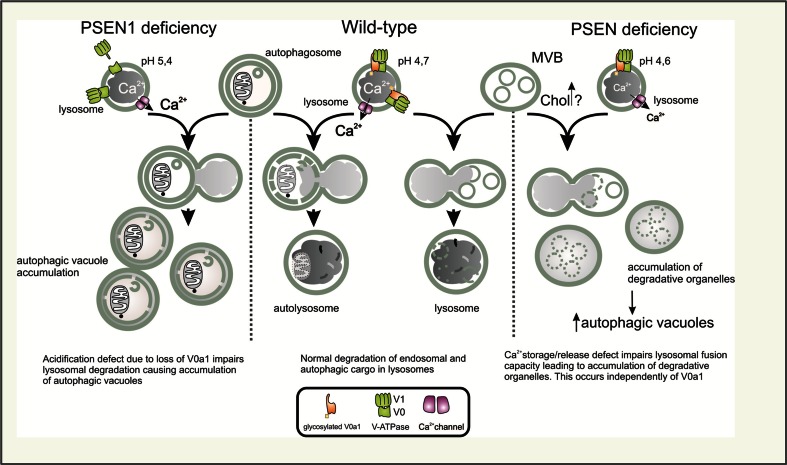
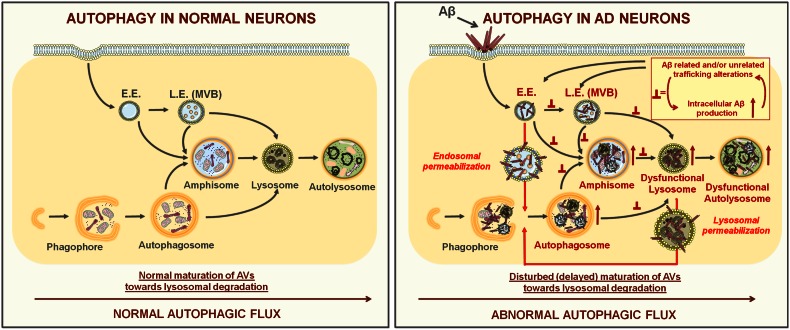
Alzheimer's disease (AD) is the most common form of dementia in the elderly. This brain neuropathology is characterized by a progressive synaptic dysfunction and neuronal loss, which lead to decline in memory and other cognitive functions. Histopathologically, AD manifests via synaptic abnormalities, neuronal degeneration as well as the deposition of extracellular amyloid plaques and intraneuronal neurofibrillary tangles. While the exact pathogenic contribution of these two AD hallmarks and their abundant constituents [aggregation-prone amyloid β (Aβ) peptide species and hyperphosphorylated tau protein, respectively] remain debated, a growing body of evidence suggests that their development may be paralleled or even preceded by the alterations/dysfunctions in the endolysosomal and the autophagic system. In AD-affected neurons, abnormalities in these cellular pathways are readily observed already at early stages of disease development, and even though many studies agree that defective lysosomal degradation may relate to or even underlie some of these deficits, specific upstream molecular defects are still deliberated. In this review we summarize various pathogenic events that may lead to these cellular abnormalities, in light of our current understanding of molecular mechanisms that govern AD progression. In addition, we also highlight the increasing evidence supporting mutual functional dependence of the endolysosomal trafficking and autophagy, in particular focusing on those molecules and processes which may be of significance to AD.
Figures
References
-
- Alonso AC, Grundke-Iqbal I, Iqbal K. Alzheimer’s disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat Med. 1996;2(7):783–787. - PubMed
-
- Annaert WG, Esselens C, Baert V, Boeve C, Snellings G, Cupers P, Craessaerts K, De Strooper B. Interaction with telencephalin and the amyloid precursor protein predicts a ring structure for presenilins. Neuron. 2001;32(4):579–589. - PubMed
-
- Aqul A, Liu B, Ramirez CM, Pieper AA, Estill SJ, Burns DK, Repa JJ, Turley SD, Dietschy JM. Unesterified cholesterol accumulation in late endosomes/lysosomes causes neurodegeneration and is prevented by driving cholesterol export from this compartment. J Neurosci. 2011;31(25):9404–9413. - PMC - PubMed
-
- Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology. 1992;42(3 Pt 1):631–639. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
