

Peroxisome proliferator-activated receptor gamma depletion stimulates Nox4 expression and human pulmonary artery smooth muscle cell proliferation
- PMID: 25557278
- PMCID: PMC4355175
- DOI: 10.1016/j.freeradbiomed.2014.12.019
Peroxisome proliferator-activated receptor gamma depletion stimulates Nox4 expression and human pulmonary artery smooth muscle cell proliferation
Abstract
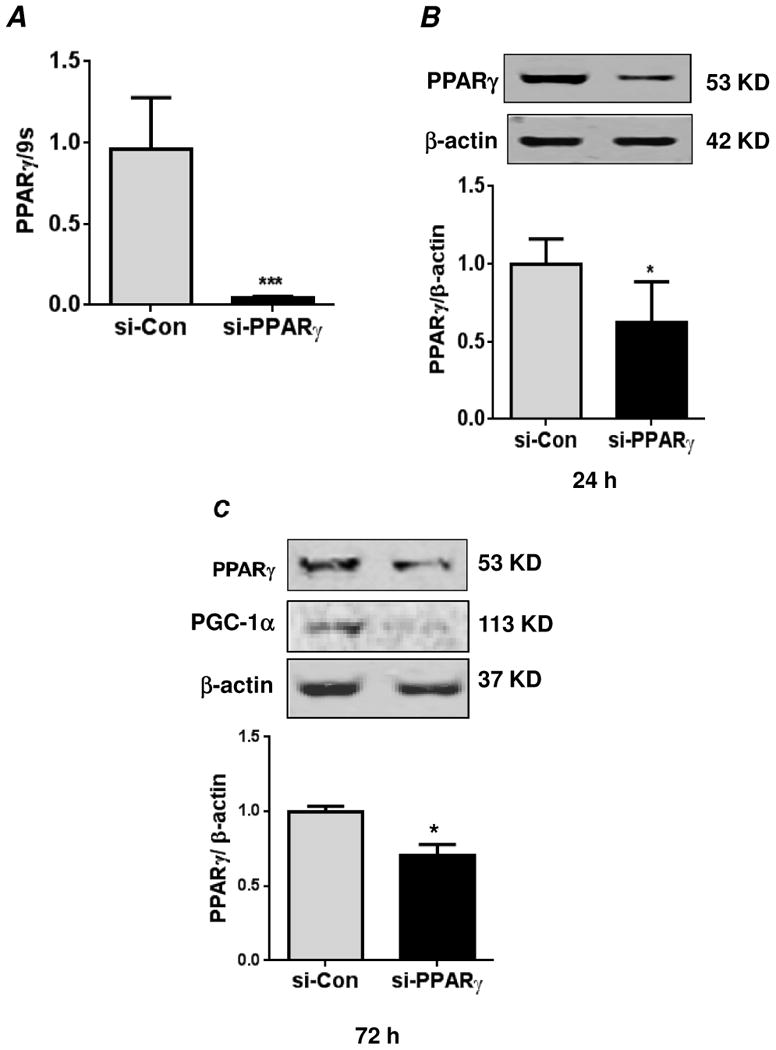
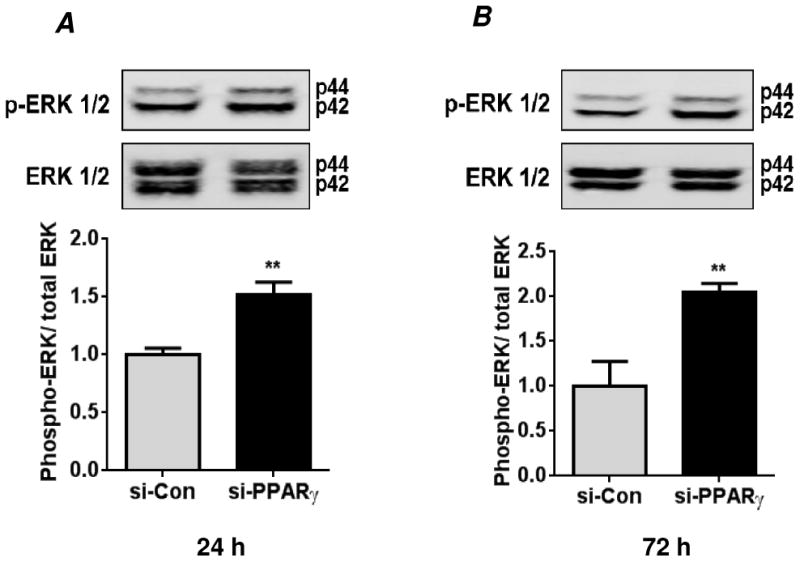
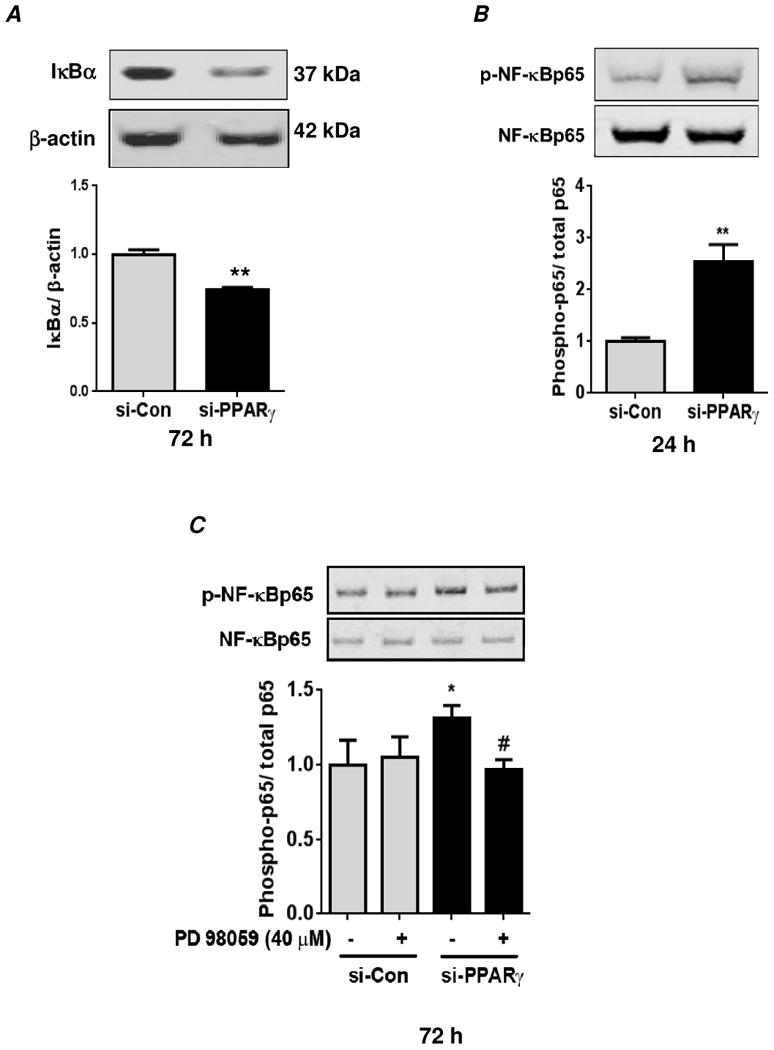
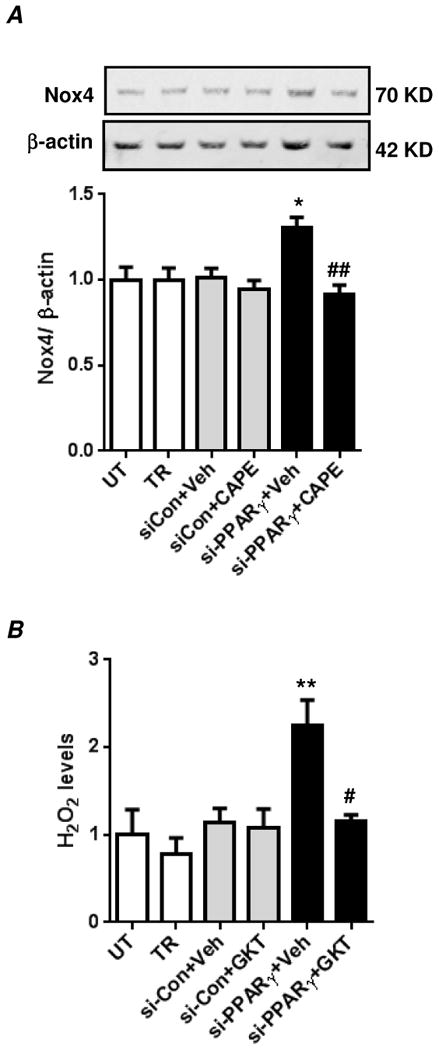
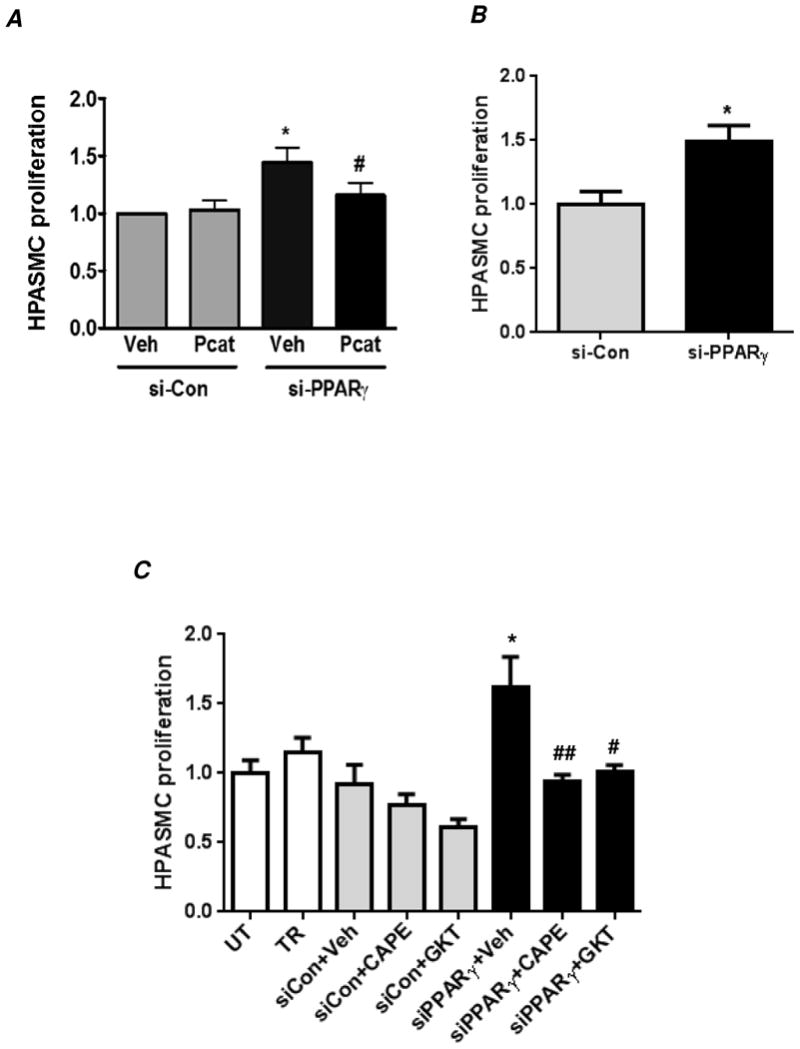
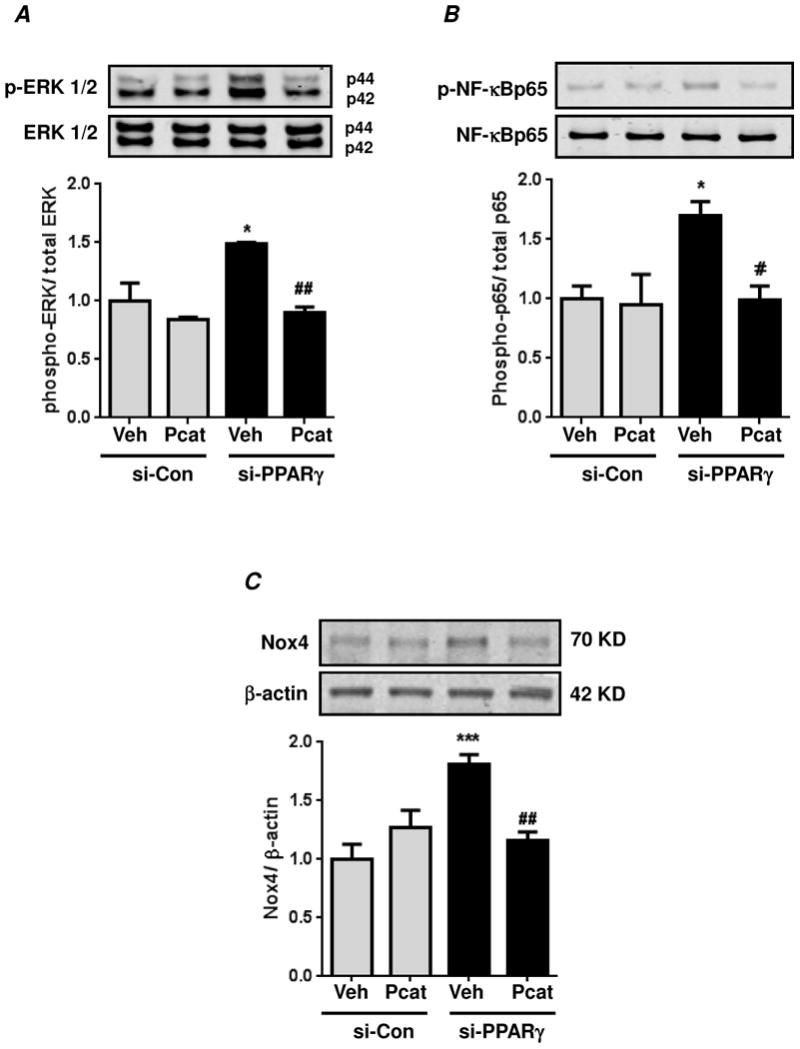
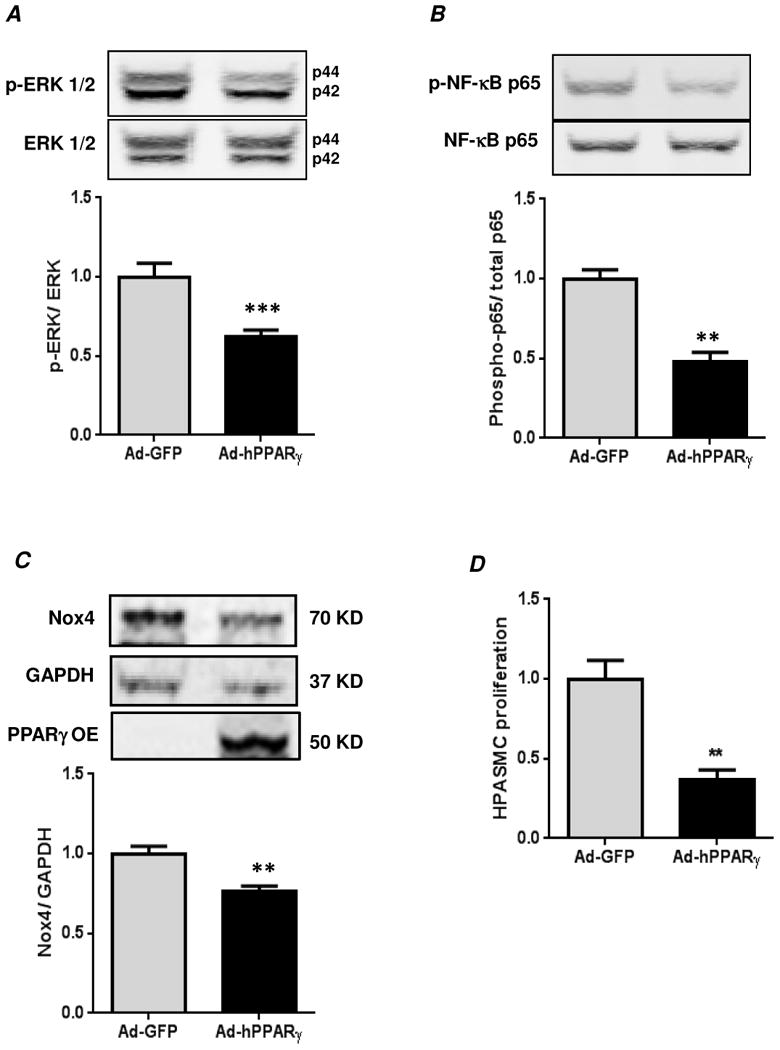
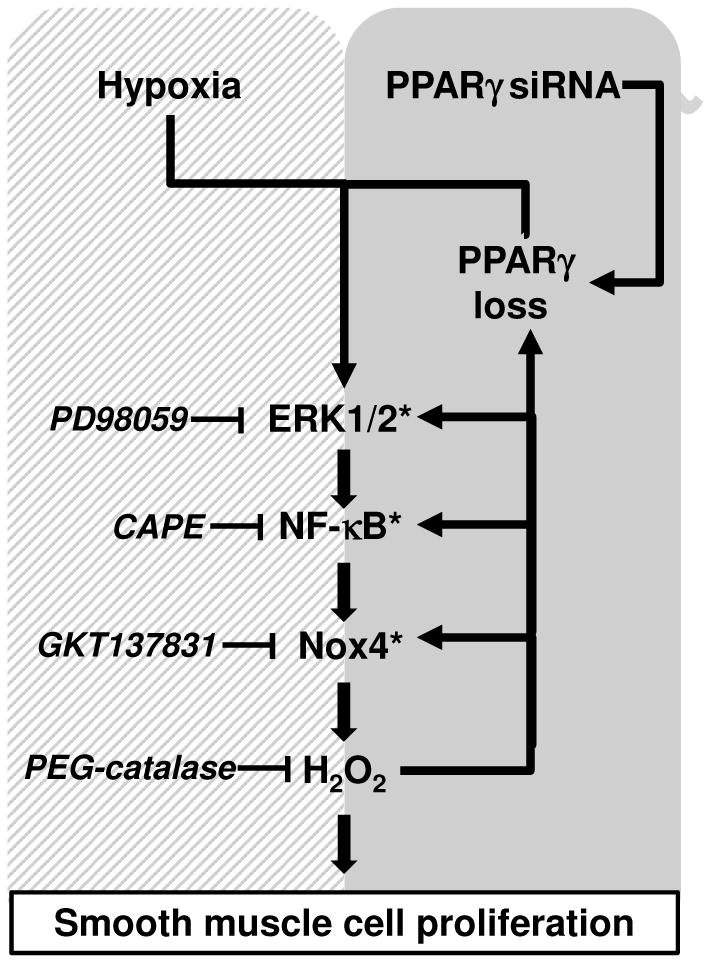
Hypoxia stimulates pulmonary hypertension (PH) in part by increasing the proliferation of pulmonary vascular wall cells. Recent evidence suggests that signaling events involved in hypoxia-induced cell proliferation include sustained nuclear factor-kappaB (NF-κB) activation, increased NADPH oxidase 4 (Nox4) expression, and downregulation of peroxisome proliferator-activated receptor gamma (PPARγ) levels. To further understand the role of reduced PPARγ levels associated with PH pathobiology, siRNA was employed to reduce PPARγ levels in human pulmonary artery smooth muscle cells (HPASMC) in vitro under normoxic conditions. PPARγ protein levels were reduced to levels comparable to those observed under hypoxic conditions. Depletion of PPARγ for 24-72 h activated mitogen-activated protein kinase, ERK 1/2, and NF-κB. Inhibition of ERK 1/2 prevented NF-κB activation caused by PPARγ depletion, indicating that ERK 1/2 lies upstream of NF-κB activation. Depletion of PPARγ for 72 h increased NF-κB-dependent Nox4 expression and H2O2 production. Inhibition of NF-κB or Nox4 attenuated PPARγ depletion-induced HPASMC proliferation. Degradation of PPARγ depletion-induced H2O2 by PEG-catalase prevented HPASMC proliferation and also ERK 1/2 and NF-κB activation and Nox4 expression, indicating that H2O2 participates in feed-forward activation of the above signaling events. Contrary to the effects of PPARγ depletion, HPASMC PPARγ overexpression reduced ERK 1/2 and NF-κB activation, Nox4 expression, and cell proliferation. Taken together these findings provide novel evidence that PPARγ plays a central role in the regulation of the ERK1/2-NF-κB-Nox4-H2O2 signaling axis in HPASMC. These results indicate that reductions in PPARγ caused by pathophysiological stimuli such as prolonged hypoxia exposure are sufficient to promote the proliferation of pulmonary vascular smooth muscle cells observed in PH pathobiology.
Keywords: ERK 1/2; NF-κB; Nox4; PPARγ; Pulmonary artery smooth muscle cell; Pulmonary hypertension.
Published by Elsevier Inc.
Figures
References
-
- Picard F, Auwerx J. PPAR(gamma) and glucose homeostasis. Annu Rev Nutr. 2002;22:167–197. - PubMed
-
- Bardot O, Aldridge TC, Latruffe N, Green S. PPAR-RXR heterodimer activates a peroxisome proliferator response element upstream of the bifunctional enzyme gene. Biochem Biophys Res Commun. 1993;192:37–45. - PubMed
-
- Tontonoz P, Hu E, Spiegelman BM. Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell. 1994;79:1147–1156. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
