

A synthetic lethal screen identifies the Vitamin D receptor as a novel gemcitabine sensitizer in pancreatic cancer cells
- PMID: 25558828
- PMCID: PMC4615005
- DOI: 10.4161/15384101.2014.967070
A synthetic lethal screen identifies the Vitamin D receptor as a novel gemcitabine sensitizer in pancreatic cancer cells
Abstract
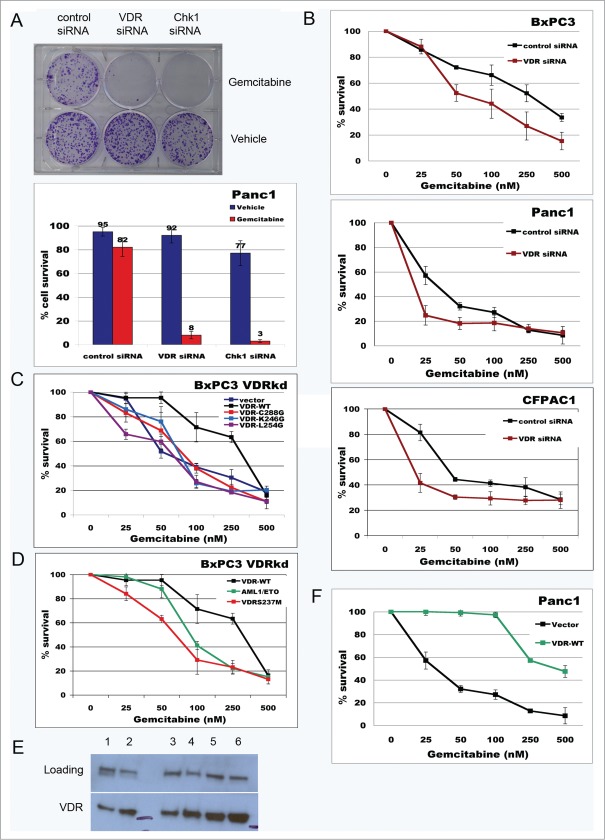
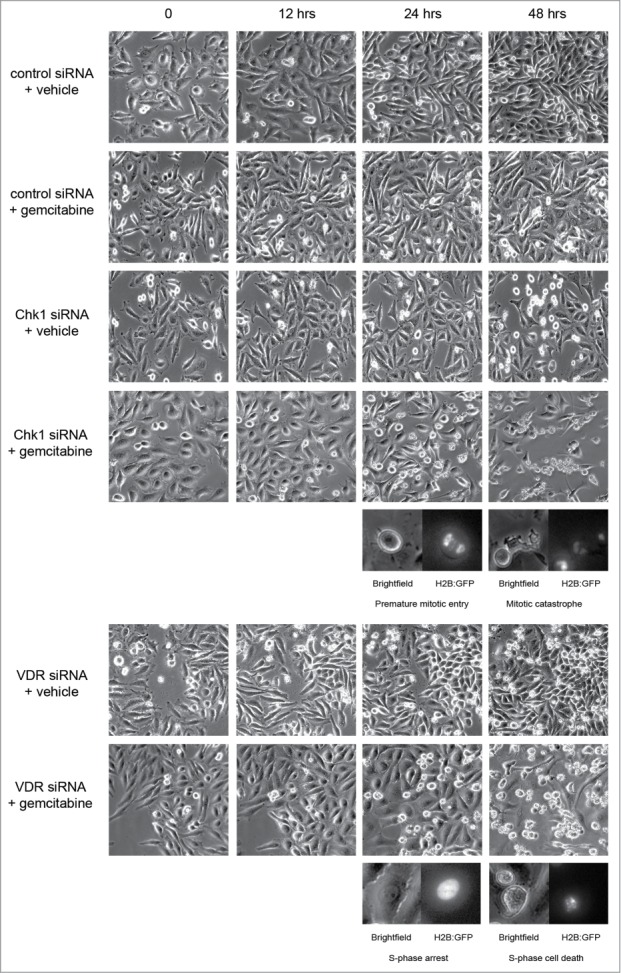
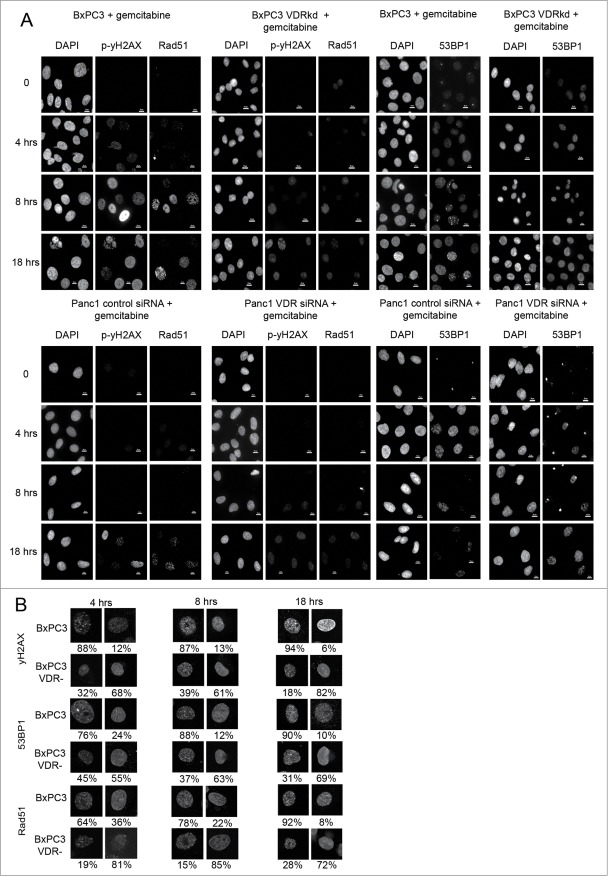
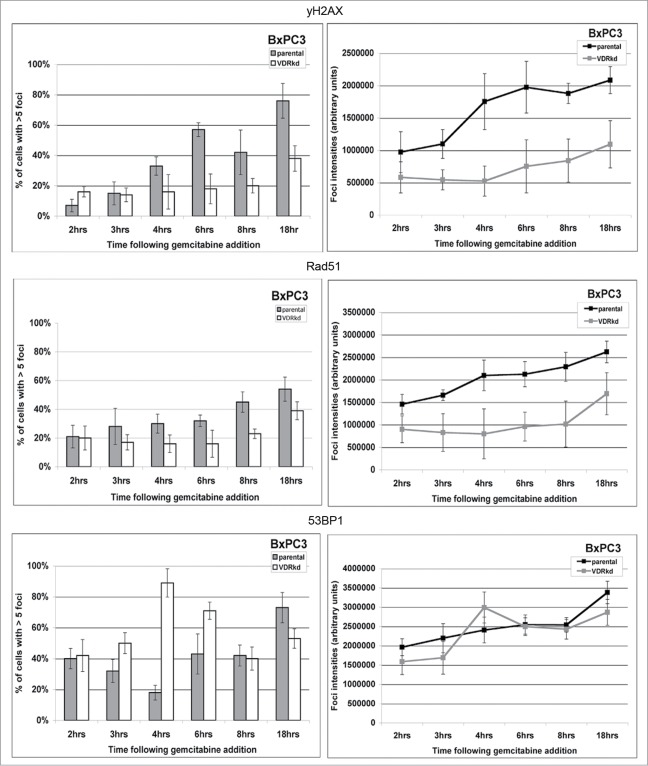
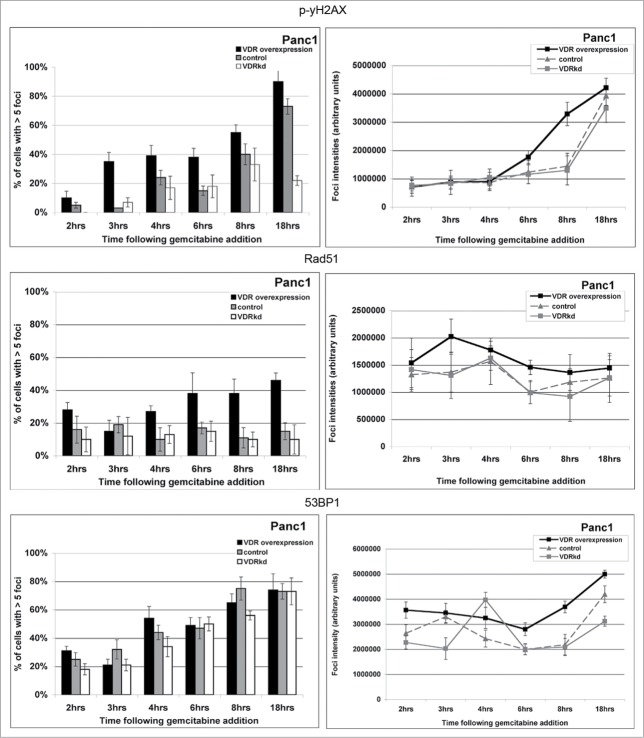
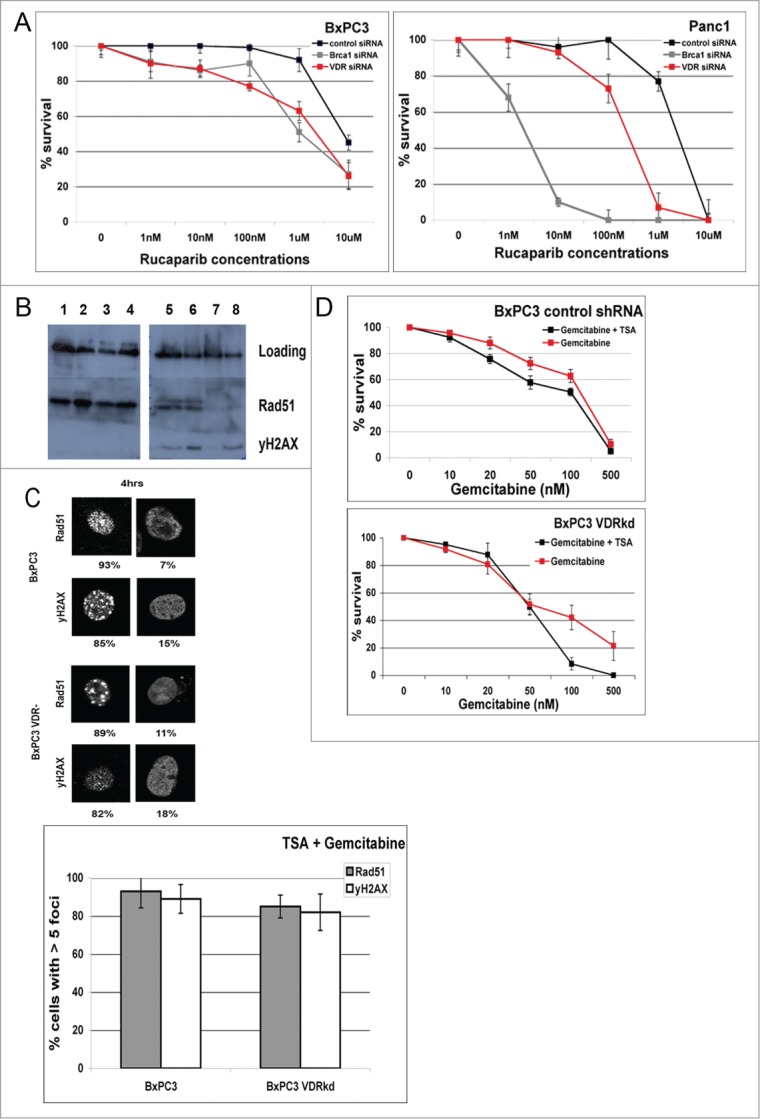
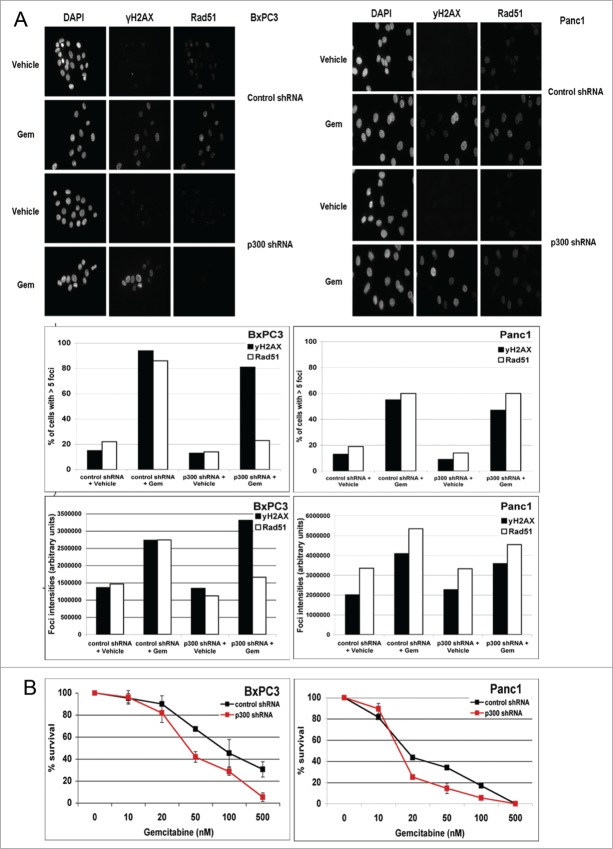
Overcoming chemoresistance of pancreatic cancer (PCa) cells should significantly extend patient survival. The current treatment modalities rely on a variety of DNA damaging agents including gemcitabine, FOLFIRINOX, and Abraxane that activate cell cycle checkpoints, which allows cells to survive these drug treaments. Indeed, these treatment regimens have only extended patient survival by a few months. The complex microenvironment of PCa tumors has been shown to complicate drug delivery thus decreasing the sensitivity of PCa tumors to chemotherapy. In this study, a genome-wide siRNA library was used to conduct a synthetic lethal screen of Panc1 cells that was treated with gemcitabine. A sublethal dose (50 nM) of the drug was used to model situations of limiting drug availability to PCa tumors in vivo. Twenty-seven validated sensitizer genes were identified from the screen including the Vitamin D receptor (VDR). Gemcitabine sensitivity was shown to be VDR dependent in multiple PCa cell lines in clonogenic survival assays. Sensitization was not achieved through checkpoint override but rather through disrupting DNA repair. VDR knockdown disrupted the cells' ability to form phospho-γH2AX and Rad51 foci in response to gemcitabine treatment. Disruption of Rad51 foci formation, which compromises homologous recombination, was consistent with increased sensitivity of PCa cells to the PARP inhibitor Rucaparib. Thus inhibition of VDR in PCa cells provides a new way to enhance the efficacy of genotoxic drugs.
Keywords: BXPC3; DNA DSB, DNA Double-strand break; DNA repair; HDAC inhibitors; IF, Immunofluorescence; PARP inhibitor; PCa, Pancreatic cancer; Panc1; Rad51 foci; VDR; VDR, Vitamin D receptor; Vitamin D receptor; chemosensitization; gemcitabine; p300; pancreatic cancer; siRNA screen; stalled replication fork.
Figures
References
-
- Caldas C, Kern SE. K-ras mutation and pancreatic adenocarcinoma. Int J Pancreatol 1995; 18:1-6; PMID:7594765 - PubMed
-
- Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008; 321:1801-6; PMID:18772397; http://dx.doi.org/ 10.1126/science.1164368 - DOI - PMC - PubMed
-
- Ko AH, Tempero MA. Treatment of metastatic pancreatic cancer. J Natl Compr Canc Netw 2005; 3:627-36; PMID:16194454 - PubMed
-
- van Heek T, Rader AE, Offerhaus GJ, McCarthy DM, Goggins M, Hruban RH, Wilentz RE. K-ras, p53, and DPC4 (MAD4) alterations in fine-needle aspirates of the pancreas: a molecular panel correlates with and supplements cytologic diagnosis. Am J Clin Pathol 2002; 117:755-65; PMID:12090425; http://dx.doi.org/ 10.1309/5RQ0-JCQU-5XF2-51LQ - DOI - PubMed
-
- Weinstein IB. Cancer. Addiction to oncogenes–the Achilles heal of cancer. Science 2002; 297:63-4; PMID:12098689; http://dx.doi.org/ 10.1126/science.1073096 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous