

MD-2 is required for disulfide HMGB1-dependent TLR4 signaling
- PMID: 25559892
- PMCID: PMC4291531
- DOI: 10.1084/jem.20141318
MD-2 is required for disulfide HMGB1-dependent TLR4 signaling
Abstract
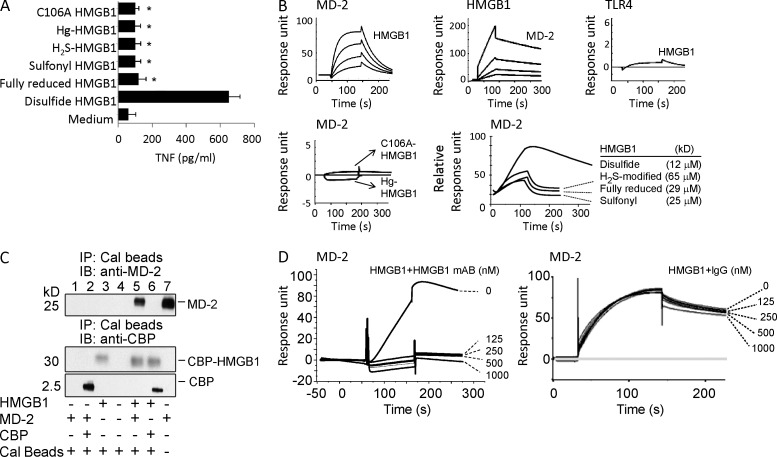
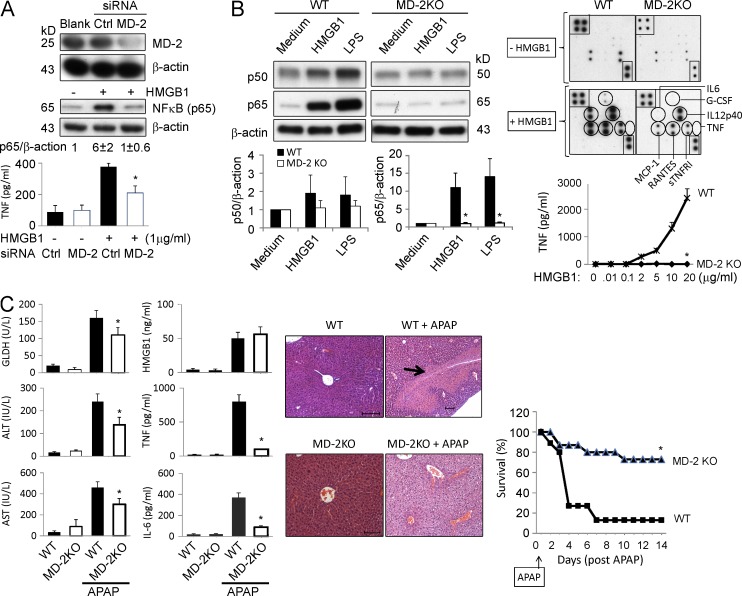
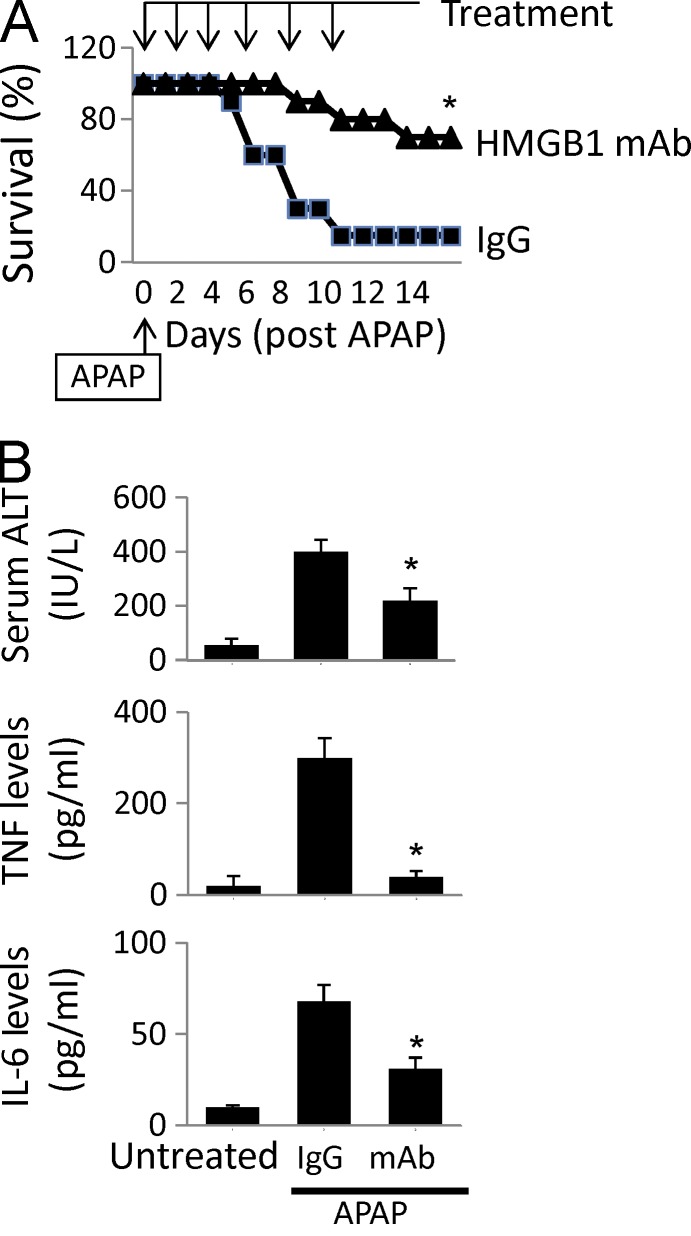
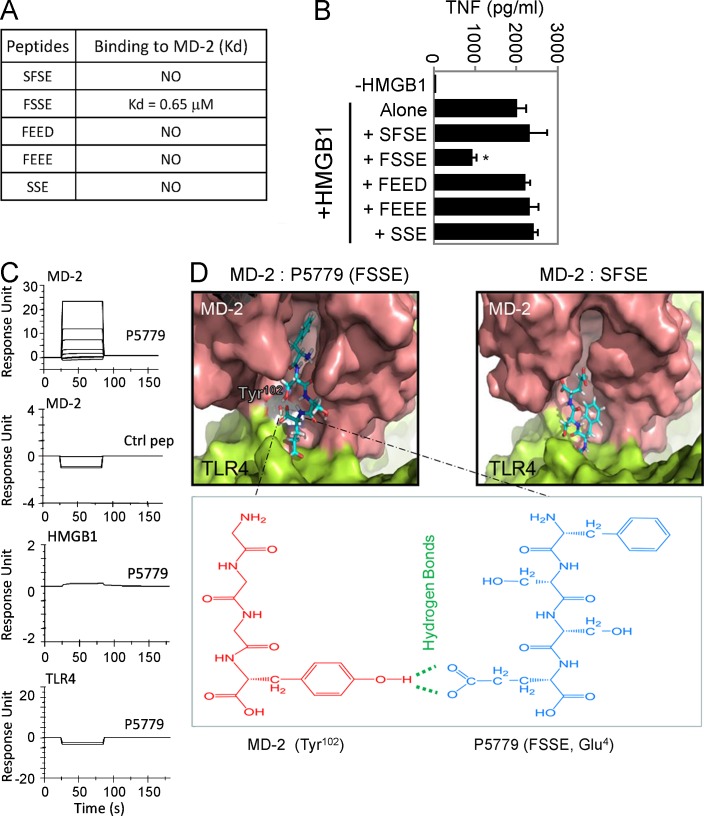
Innate immune receptors for pathogen- and damage-associated molecular patterns (PAMPs and DAMPs) orchestrate inflammatory responses to infection and injury. Secreted by activated immune cells or passively released by damaged cells, HMGB1 is subjected to redox modification that distinctly influences its extracellular functions. Previously, it was unknown how the TLR4 signalosome distinguished between HMGB1 isoforms. Here we demonstrate that the extracellular TLR4 adaptor, myeloid differentiation factor 2 (MD-2), binds specifically to the cytokine-inducing disulfide isoform of HMGB1, to the exclusion of other isoforms. Using MD-2-deficient mice, as well as MD-2 silencing in macrophages, we show a requirement for HMGB1-dependent TLR4 signaling. By screening HMGB1 peptide libraries, we identified a tetramer (FSSE, designated P5779) as a specific MD-2 antagonist preventing MD-2-HMGB1 interaction and TLR4 signaling. P5779 does not interfere with lipopolysaccharide-induced cytokine/chemokine production, thus preserving PAMP-mediated TLR4-MD-2 responses. Furthermore, P5779 can protect mice against hepatic ischemia/reperfusion injury, chemical toxicity, and sepsis. These findings reveal a novel mechanism by which innate systems selectively recognize specific HMGB1 isoforms. The results may direct toward strategies aimed at attenuating DAMP-mediated inflammation while preserving antimicrobial immune responsiveness.
© 2015 Yang et al.
Figures
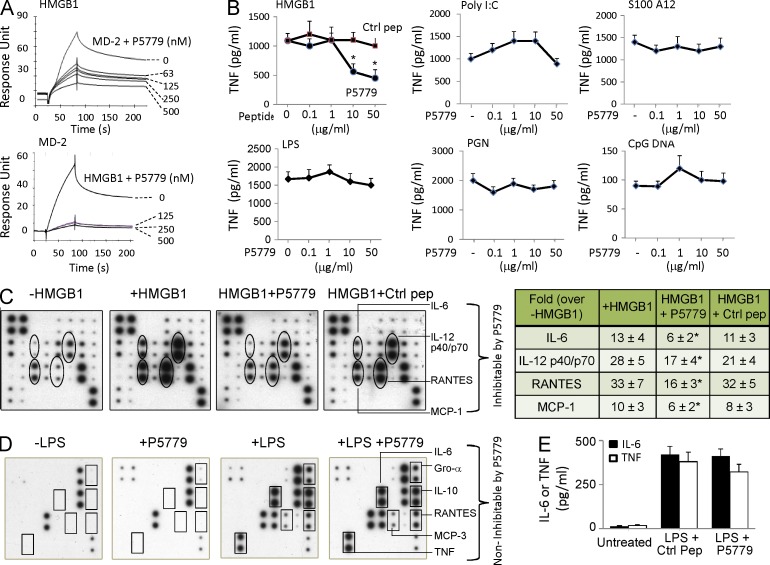
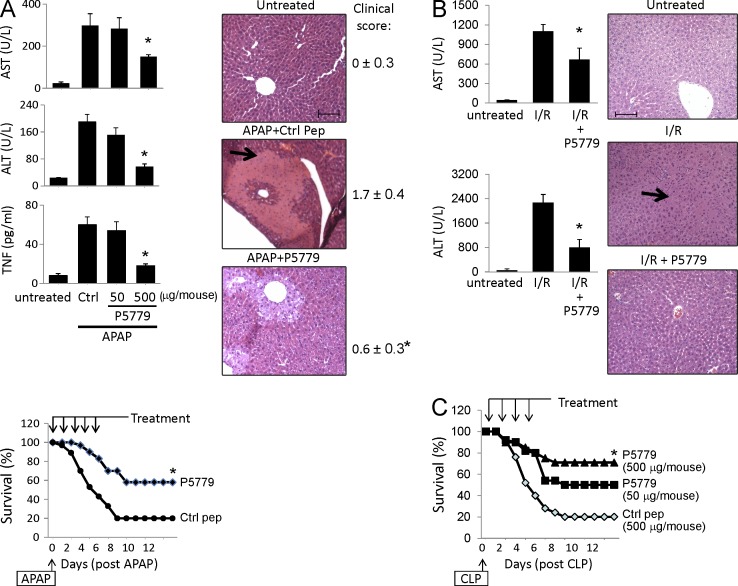
Similar articles
-
Folic acid derived-P5779 mimetics regulate DAMP-mediated inflammation through disruption of HMGB1:TLR4:MD-2 axes.PLoS One. 2018 Feb 15;13(2):e0193028. doi: 10.1371/journal.pone.0193028. eCollection 2018. PLoS One. 2018. PMID: 29447234 Free PMC article.
-
A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release.Proc Natl Acad Sci U S A. 2010 Jun 29;107(26):11942-7. doi: 10.1073/pnas.1003893107. Epub 2010 Jun 14. Proc Natl Acad Sci U S A. 2010. PMID: 20547845 Free PMC article.
-
Signaling of high mobility group box 1 (HMGB1) through toll-like receptor 4 in macrophages requires CD14.Mol Med. 2013 May 20;19(1):88-98. doi: 10.2119/molmed.2012.00306. Mol Med. 2013. PMID: 23508573 Free PMC article.
-
Targeting Inflammation Driven by HMGB1.Front Immunol. 2020 Mar 20;11:484. doi: 10.3389/fimmu.2020.00484. eCollection 2020. Front Immunol. 2020. PMID: 32265930 Free PMC article. Review.
-
Drug Targeting Based on a New Concept-Targeting Against TLR4 as an Example.Endocr Metab Immune Disord Drug Targets. 2015;15(2):83-7. doi: 10.2174/187153031502150522123746. Endocr Metab Immune Disord Drug Targets. 2015. PMID: 26004773 Review.
Cited by
-
Targeting HMGB1 for the treatment of sepsis and sepsis-induced organ injury.Acta Pharmacol Sin. 2022 Mar;43(3):520-528. doi: 10.1038/s41401-021-00676-7. Epub 2021 May 26. Acta Pharmacol Sin. 2022. PMID: 34040166 Free PMC article. Review.
-
HMGB1 Activates Proinflammatory Signaling via TLR5 Leading to Allodynia.Cell Rep. 2016 Oct 18;17(4):1128-1140. doi: 10.1016/j.celrep.2016.09.076. Cell Rep. 2016. PMID: 27760316 Free PMC article.
-
Therapeutic Potential of Targeting the HMGB1/RAGE Axis in Inflammatory Diseases.Molecules. 2022 Oct 27;27(21):7311. doi: 10.3390/molecules27217311. Molecules. 2022. PMID: 36364135 Free PMC article. Review.
-
HMGB1 as a therapeutic target in disease.J Cell Physiol. 2021 May;236(5):3406-3419. doi: 10.1002/jcp.30125. Epub 2020 Oct 26. J Cell Physiol. 2021. PMID: 33107103 Free PMC article. Review.
-
ATP/P2X7 receptor signaling as a potential anti-inflammatory target of natural polyphenols.PLoS One. 2018 Sep 24;13(9):e0204229. doi: 10.1371/journal.pone.0204229. eCollection 2018. PLoS One. 2018. PMID: 30248132 Free PMC article.
References
-
- Antoine D.J., Jenkins R.E., Dear J.W., Williams D.P., McGill M.R., Sharpe M.R., Craig D.G., Simpson K.J., Jaeschke H., and Park B.K.. 2012. Molecular forms of HMGB1 and keratin-18 as mechanistic biomarkers for mode of cell death and prognosis during clinical acetaminophen hepatotoxicity. J. Hepatol. 56:1070–1079 10.1016/j.jhep.2011.12.019 - DOI - PMC - PubMed
-
- Antoine D.J., Dear J.W., Lewis P.S., Platt V., Coyle J., Masson M., Thanacoody R.H., Gray A.J., Webb D.J., Moggs J.G., et al. . 2013. Mechanistic biomarkers provide early and sensitive detection of acetaminophen-induced acute liver injury at first presentation to hospital. Hepatology. 58:777–787 10.1002/hep.26294 - DOI - PMC - PubMed
-
- Deng M., Scott M.J., Loughran P., Gibson G., Sodhi C., Watkins S., Hackam D., and Billiar T.R.. 2013. Lipopolysaccharide clearance, bacterial clearance, and systemic inflammatory responses are regulated by cell type-specific functions of TLR4 during sepsis. J. Immunol. 190:5152–5160 10.4049/jimmunol.1300496 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
- R01 GM106314/GM/NIGMS NIH HHS/United States
- M01 RR018535/RR/NCRR NIH HHS/United States
- R01 GM062508/GM/NIGMS NIH HHS/United States
- R37 GM054060/GM/NIGMS NIH HHS/United States
- R01 AT005076/AT/NCCIH NIH HHS/United States
- P50 GM053789/GM/NIGMS NIH HHS/United States
- R01 GM098446/GM/NIGMS NIH HHS/United States
- R01GM107876/GM/NIGMS NIH HHS/United States
- R01GM62508/GM/NIGMS NIH HHS/United States
- R01 GM107846/GM/NIGMS NIH HHS/United States
- MR/L006758/1/MRC_/Medical Research Council/United Kingdom
- 5P50GM053789/GM/NIGMS NIH HHS/United States
- M01RR018535/RR/NCRR NIH HHS/United States
- R01GM098446/GM/NIGMS NIH HHS/United States
- R01AT005076/AT/NCCIH NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
