

BRF1 mutations alter RNA polymerase III-dependent transcription and cause neurodevelopmental anomalies
- PMID: 25561519
- PMCID: PMC4315290
- DOI: 10.1101/gr.176925.114
BRF1 mutations alter RNA polymerase III-dependent transcription and cause neurodevelopmental anomalies
Erratum in
-
BRF1 mutations alter RNA polymerase III-dependent transcription and cause neurodevelopmental anomalies.Genome Res. 2015 Apr;25(4):609. Genome Res. 2015. PMID: 25834187 Free PMC article. No abstract available.
Abstract
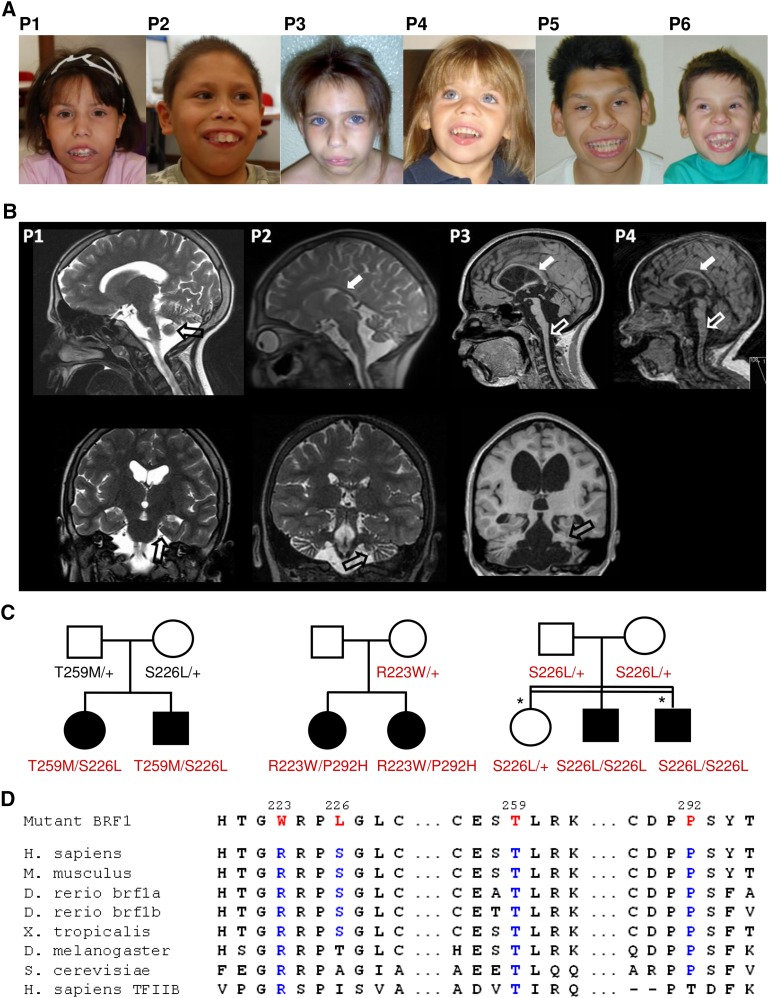
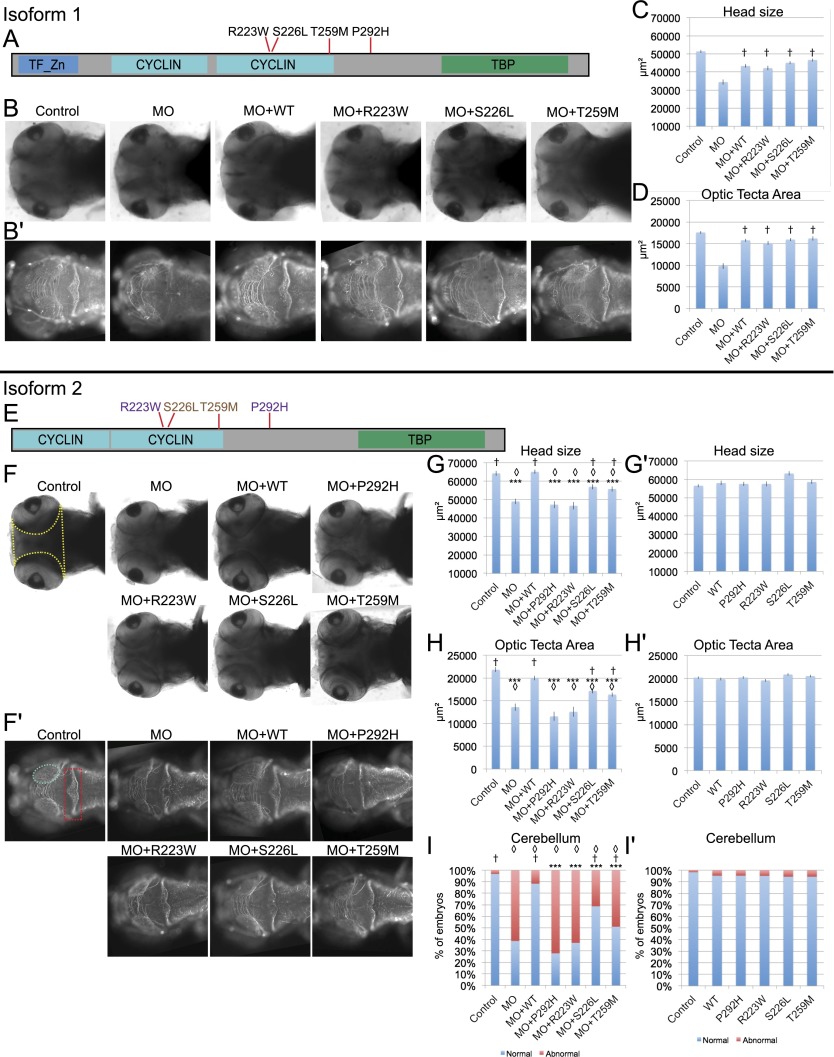
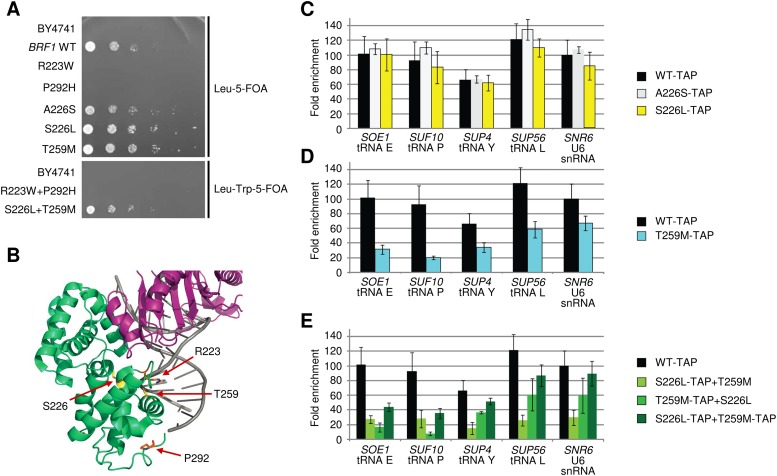
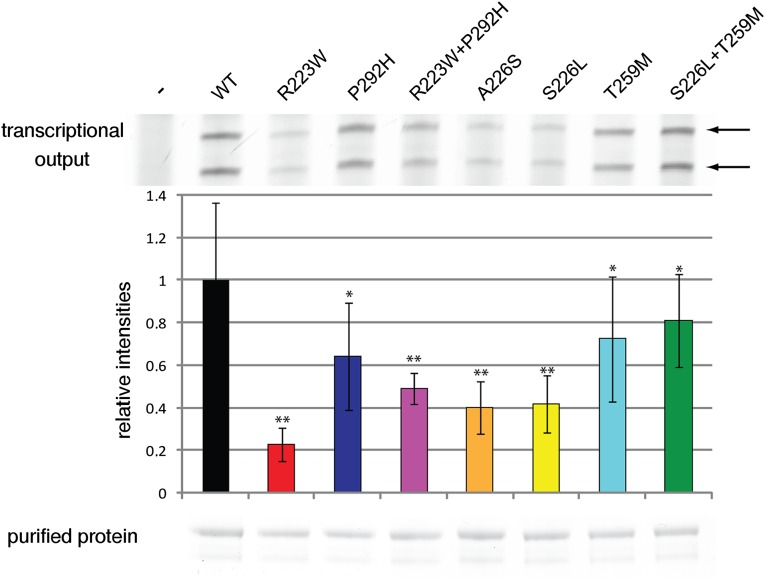
RNA polymerase III (Pol III) synthesizes tRNAs and other small noncoding RNAs to regulate protein synthesis. Dysregulation of Pol III transcription has been linked to cancer, and germline mutations in genes encoding Pol III subunits or tRNA processing factors cause neurogenetic disorders in humans, such as hypomyelinating leukodystrophies and pontocerebellar hypoplasia. Here we describe an autosomal recessive disorder characterized by cerebellar hypoplasia and intellectual disability, as well as facial dysmorphic features, short stature, microcephaly, and dental anomalies. Whole-exome sequencing revealed biallelic missense alterations of BRF1 in three families. In support of the pathogenic potential of the discovered alleles, suppression or CRISPR-mediated deletion of brf1 in zebrafish embryos recapitulated key neurodevelopmental phenotypes; in vivo complementation showed all four candidate mutations to be pathogenic in an apparent isoform-specific context. BRF1 associates with BDP1 and TBP to form the transcription factor IIIB (TFIIIB), which recruits Pol III to target genes. We show that disease-causing mutations reduce Brf1 occupancy at tRNA target genes in Saccharomyces cerevisiae and impair cell growth. Moreover, BRF1 mutations reduce Pol III-related transcription activity in vitro. Taken together, our data show that BRF1 mutations that reduce protein activity cause neurodevelopmental anomalies, suggesting that BRF1-mediated Pol III transcription is required for normal cerebellar and cognitive development.
© 2015 Borck et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
Similar articles
-
Expanding the phenotype of cerebellar-facial-dental syndrome: Two siblings with a novel variant in BRF1.Am J Med Genet A. 2020 Nov;182(11):2742-2745. doi: 10.1002/ajmg.a.61839. Epub 2020 Sep 8. Am J Med Genet A. 2020. PMID: 32896090
-
Differential expression of the TFIIIB subunits Brf1 and Brf2 in cancer cells.BMC Mol Biol. 2008 Aug 12;9:74. doi: 10.1186/1471-2199-9-74. BMC Mol Biol. 2008. PMID: 18700021 Free PMC article.
-
RNF12 catalyzes BRF1 ubiquitination and regulates RNA polymerase III-dependent transcription.J Biol Chem. 2019 Jan 4;294(1):130-141. doi: 10.1074/jbc.RA118.004524. Epub 2018 Nov 9. J Biol Chem. 2019. PMID: 30413534 Free PMC article.
-
ROS Signaling-Mediated Novel Biological Targets: Brf1 and RNA Pol III Genes.Oxid Med Cell Longev. 2021 Oct 4;2021:5888432. doi: 10.1155/2021/5888432. eCollection 2021. Oxid Med Cell Longev. 2021. Retraction in: Oxid Med Cell Longev. 2024 Jan 9;2024:9890237. doi: 10.1155/2024/9890237. PMID: 34646425 Free PMC article. Retracted. Review.
-
SMARCE1, a rare cause of Coffin-Siris Syndrome: Clinical description of three additional cases.Am J Med Genet A. 2016 Aug;170(8):1967-73. doi: 10.1002/ajmg.a.37722. Epub 2016 Jun 5. Am J Med Genet A. 2016. PMID: 27264197 Free PMC article. Review.
Cited by
-
Bi-allelic Variants in DYNC1I2 Cause Syndromic Microcephaly with Intellectual Disability, Cerebral Malformations, and Dysmorphic Facial Features.Am J Hum Genet. 2019 Jun 6;104(6):1073-1087. doi: 10.1016/j.ajhg.2019.04.002. Epub 2019 May 9. Am J Hum Genet. 2019. PMID: 31079899 Free PMC article.
-
tRNA Metabolism and Neurodevelopmental Disorders.Annu Rev Genomics Hum Genet. 2019 Aug 31;20:359-387. doi: 10.1146/annurev-genom-083118-015334. Epub 2019 May 13. Annu Rev Genomics Hum Genet. 2019. PMID: 31082281 Free PMC article. Review.
-
From APC to the genetics of hereditary and familial colon cancer syndromes.Hum Mol Genet. 2021 Oct 1;30(R2):R206-R224. doi: 10.1093/hmg/ddab208. Hum Mol Genet. 2021. PMID: 34329396 Free PMC article. Review.
-
Chromatin Alterations in Neurological Disorders and Strategies of (Epi)Genome Rescue.Pharmaceuticals (Basel). 2021 Aug 4;14(8):765. doi: 10.3390/ph14080765. Pharmaceuticals (Basel). 2021. PMID: 34451862 Free PMC article. Review.
-
Whole-exome sequencing identified a novel variant in an Iranian patient affected by pycnodysostosis.Mol Genet Genomic Med. 2020 Mar;8(3):e1118. doi: 10.1002/mgg3.1118. Epub 2020 Jan 15. Mol Genet Genomic Med. 2020. PMID: 31944631 Free PMC article.
References
-
- Alexander DE, Kaczorowski DJ, Jackson-Fisher AJ, Lowery DM, Zanton SJ, Pugh BF. 2004. Inhibition of TATA binding protein dimerization by RNA polymerase III transcription initiation factor Brf1. J Biol Chem 279: 32401–32406. - PubMed
-
- Alfaiz AA, Micale L, Mandriani B, Augello B, Pellico MT, Chrast J, Xenarios I, Zelante L, Merla G, Reymond A. 2014. TBC1D7 mutations are associated with intellectual disability, macrocrania, patellar dislocation, and celiac disease. Hum Mutat 35: 447–451. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases