

Efficient inference of population size histories and locus-specific mutation rates from large-sample genomic variation data
- PMID: 25564017
- PMCID: PMC4315300
- DOI: 10.1101/gr.178756.114
Efficient inference of population size histories and locus-specific mutation rates from large-sample genomic variation data
Abstract
With the recent increase in study sample sizes in human genetics, there has been growing interest in inferring historical population demography from genomic variation data. Here, we present an efficient inference method that can scale up to very large samples, with tens or hundreds of thousands of individuals. Specifically, by utilizing analytic results on the expected frequency spectrum under the coalescent and by leveraging the technique of automatic differentiation, which allows us to compute gradients exactly, we develop a very efficient algorithm to infer piecewise-exponential models of the historical effective population size from the distribution of sample allele frequencies. Our method is orders of magnitude faster than previous demographic inference methods based on the frequency spectrum. In addition to inferring demography, our method can also accurately estimate locus-specific mutation rates. We perform extensive validation of our method on simulated data and show that it can accurately infer multiple recent epochs of rapid exponential growth, a signal that is difficult to pick up with small sample sizes. Lastly, we use our method to analyze data from recent sequencing studies, including a large-sample exome-sequencing data set of tens of thousands of individuals assayed at a few hundred genic regions.
© 2015 Bhaskar et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
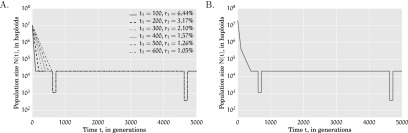
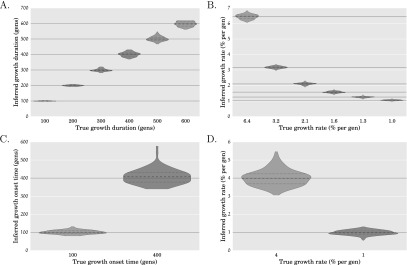
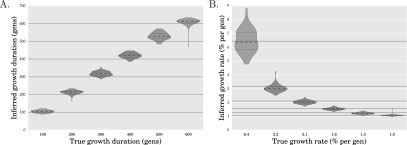
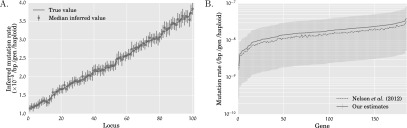
References
-
- Balding DJ, Nichols RA. 1997. Significant genetic correlations among Caucasians at forensic DNA loci. Heredity 78: 583–589. - PubMed
-
- Campbell CD, Ogburn EL, Lunetta KL, Lyon HN, Freedman ML, Groop LC, Altshuler D, Ardlie KG, Hirschhorn JN. 2005. Demonstrating stratification in a European American population. Nat Genet 37: 868–872. - PubMed
-
- Chen H. 2012. The joint allele frequency spectrum of multiple populations: a coalescent theory approach. Theor Popul Biol 81: 179–195. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources