

Hypermutation in human cancer genomes: footprints and mechanisms
- PMID: 25568919
- PMCID: PMC4280484
- DOI: 10.1038/nrc3816
Hypermutation in human cancer genomes: footprints and mechanisms
Erratum in
- Nat Rev Cancer. 2015 Nov;15(11):694
Abstract
A role for somatic mutations in carcinogenesis is well accepted, but the degree to which mutation rates influence cancer initiation and development is under continuous debate. Recently accumulated genomic data have revealed that thousands of tumour samples are riddled by hypermutation, broadening support for the idea that many cancers acquire a mutator phenotype. This major expansion of cancer mutation data sets has provided unprecedented statistical power for the analysis of mutation spectra, which has confirmed several classical sources of mutation in cancer, highlighted new prominent mutation sources (such as apolipoprotein B mRNA editing enzyme catalytic polypeptide-like (APOBEC) enzymes) and empowered the search for cancer drivers. The confluence of cancer mutation genomics and mechanistic insight provides great promise for understanding the basic development of cancer through mutations.
Figures
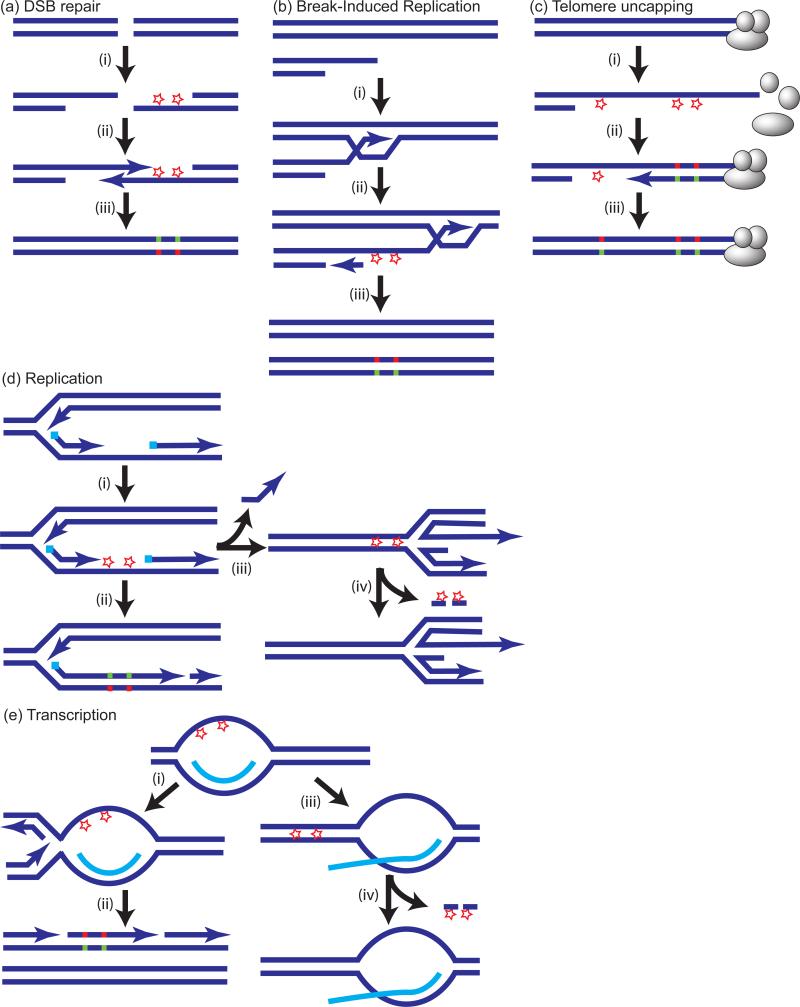
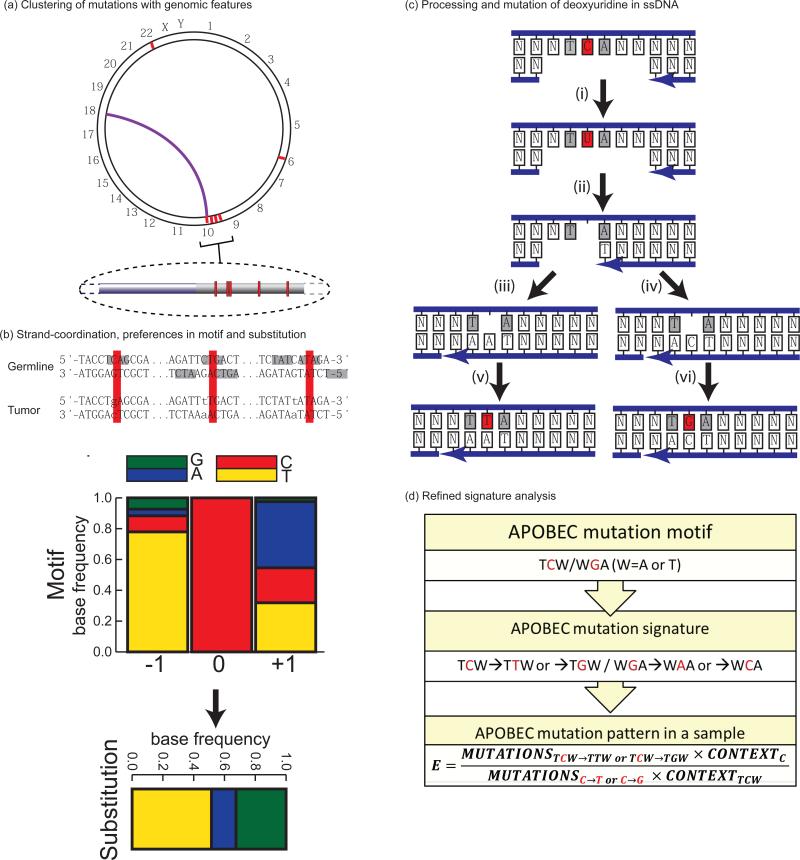
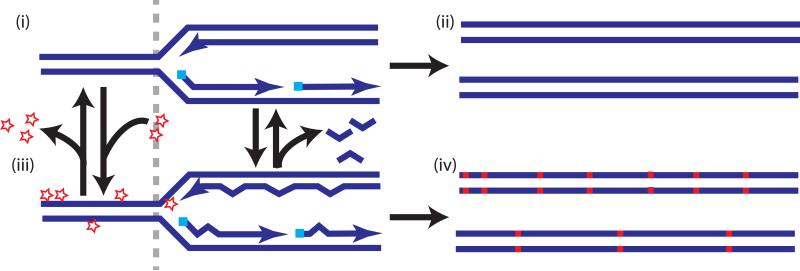
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
