

Systems level analysis of systemic sclerosis shows a network of immune and profibrotic pathways connected with genetic polymorphisms
- PMID: 25569146
- PMCID: PMC4288710
- DOI: 10.1371/journal.pcbi.1004005
Systems level analysis of systemic sclerosis shows a network of immune and profibrotic pathways connected with genetic polymorphisms
Erratum in
-
Correction: Systems level analysis of systemic sclerosis shows a network of immune and profibrotic pathways connected with genetic polymorphisms.PLoS Comput Biol. 2015 Mar 5;11(3):e1004133. doi: 10.1371/journal.pcbi.1004133. eCollection 2015 Mar. PLoS Comput Biol. 2015. PMID: 25742131 Free PMC article. No abstract available.
Abstract
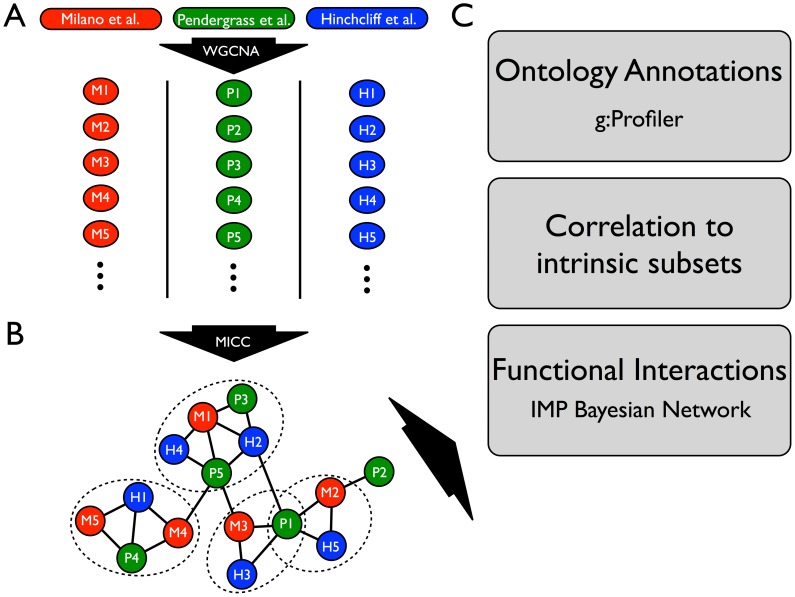
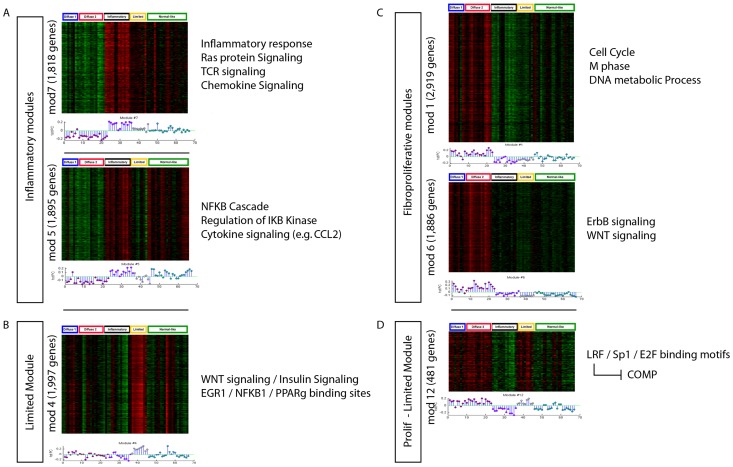
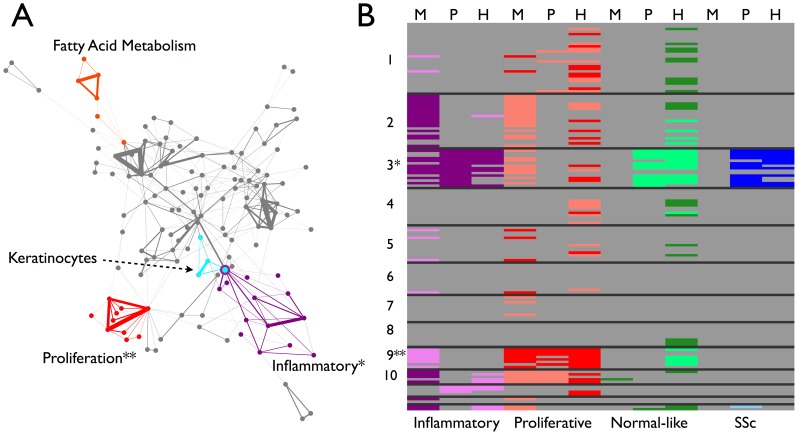
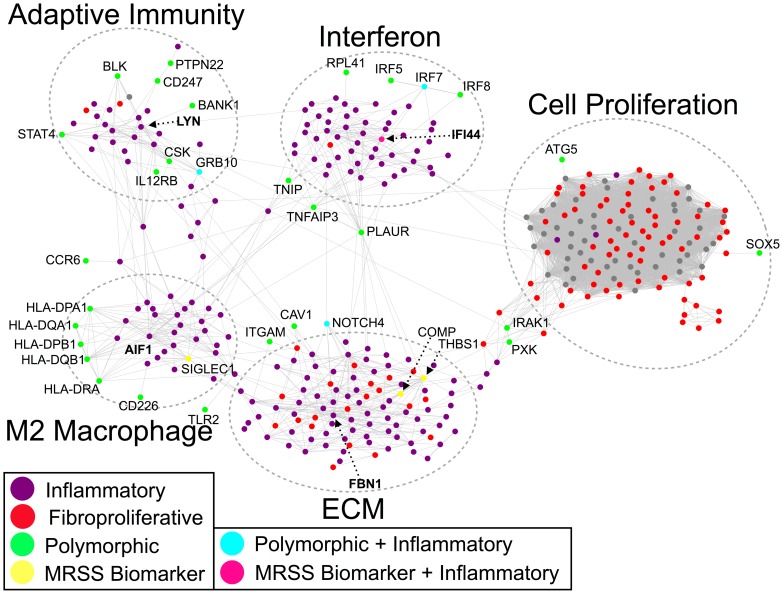
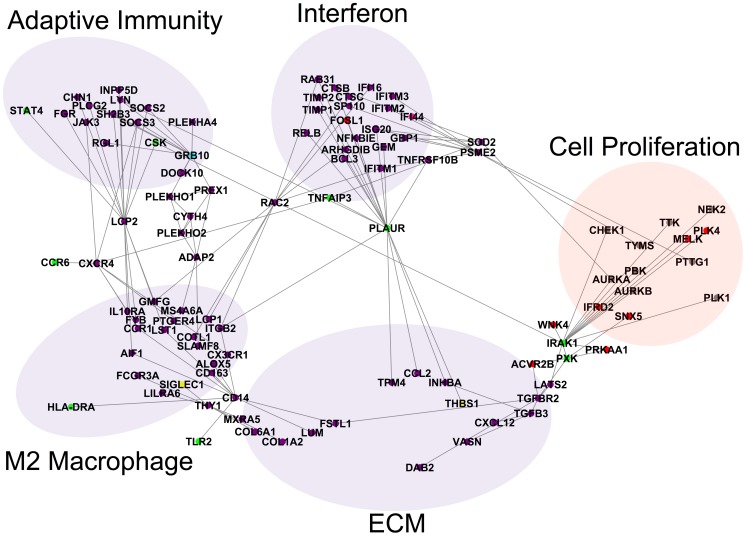
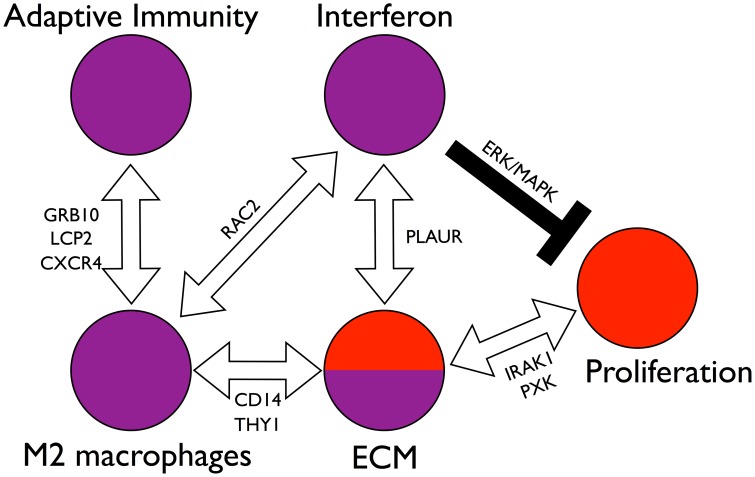
Systemic sclerosis (SSc) is a rare systemic autoimmune disease characterized by skin and organ fibrosis. The pathogenesis of SSc and its progression are poorly understood. The SSc intrinsic gene expression subsets (inflammatory, fibroproliferative, normal-like, and limited) are observed in multiple clinical cohorts of patients with SSc. Analysis of longitudinal skin biopsies suggests that a patient's subset assignment is stable over 6-12 months. Genetically, SSc is multi-factorial with many genetic risk loci for SSc generally and for specific clinical manifestations. Here we identify the genes consistently associated with the intrinsic subsets across three independent cohorts, show the relationship between these genes using a gene-gene interaction network, and place the genetic risk loci in the context of the intrinsic subsets. To identify gene expression modules common to three independent datasets from three different clinical centers, we developed a consensus clustering procedure based on mutual information of partitions, an information theory concept, and performed a meta-analysis of these genome-wide gene expression datasets. We created a gene-gene interaction network of the conserved molecular features across the intrinsic subsets and analyzed their connections with SSc-associated genetic polymorphisms. The network is composed of distinct, but interconnected, components related to interferon activation, M2 macrophages, adaptive immunity, extracellular matrix remodeling, and cell proliferation. The network shows extensive connections between the inflammatory- and fibroproliferative-specific genes. The network also shows connections between these subset-specific genes and 30 SSc-associated polymorphic genes including STAT4, BLK, IRF7, NOTCH4, PLAUR, CSK, IRAK1, and several human leukocyte antigen (HLA) genes. Our analyses suggest that the gene expression changes underlying the SSc subsets may be long-lived, but mechanistically interconnected and related to a patients underlying genetic risk.
Conflict of interest statement
I have read the journal's policy and have the following conflicts: MLW and MEH have filed patents for gene expression biomarkers in SSc. MLW is the Scientific Founder of Celdara Medical LLC.
Figures

References
-
- Whitfield ML, George LK, Grant GD, Perou CM (2006) Common markers of proliferation. Nat Rev Cancer 6: 99–106. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
