

HIF2α signaling inhibits adherens junctional disruption in acute lung injury
- PMID: 25574837
- PMCID: PMC4319409
- DOI: 10.1172/JCI77701
HIF2α signaling inhibits adherens junctional disruption in acute lung injury
Erratum in
-
Corrigendum. HIF2α signaling inhibits adherens junctional disruption in acute lung injury.J Clin Invest. 2015 Mar 2;125(3):1364. doi: 10.1172/JCI81377. Epub 2015 Mar 2. J Clin Invest. 2015. PMID: 25729855 Free PMC article. No abstract available.
Abstract
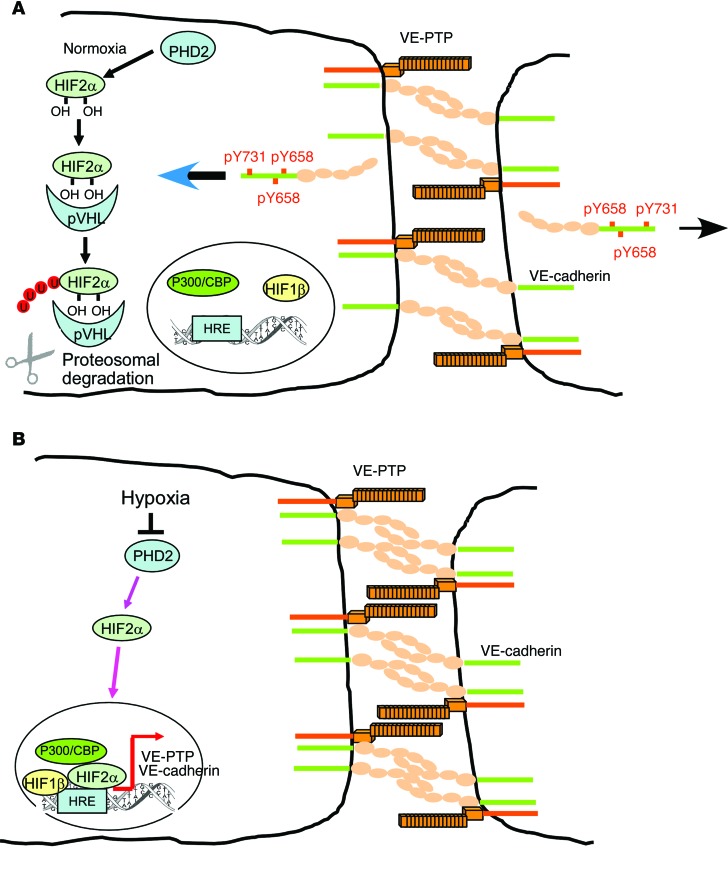
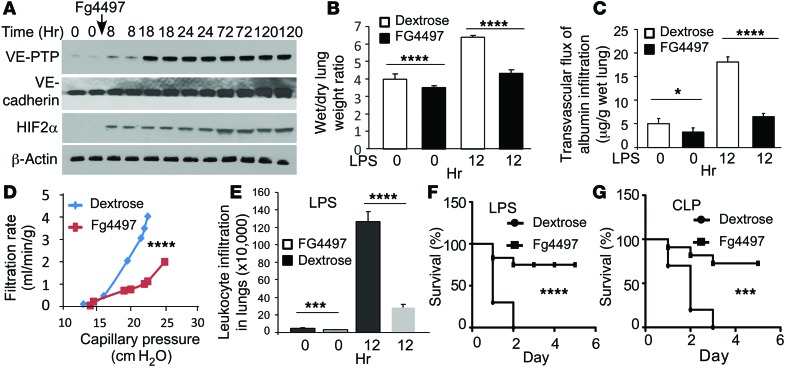
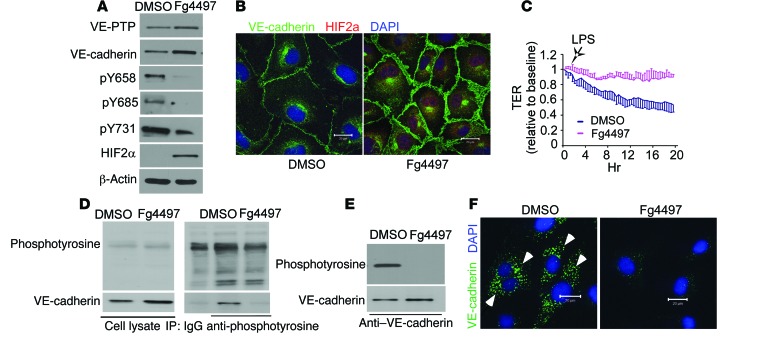
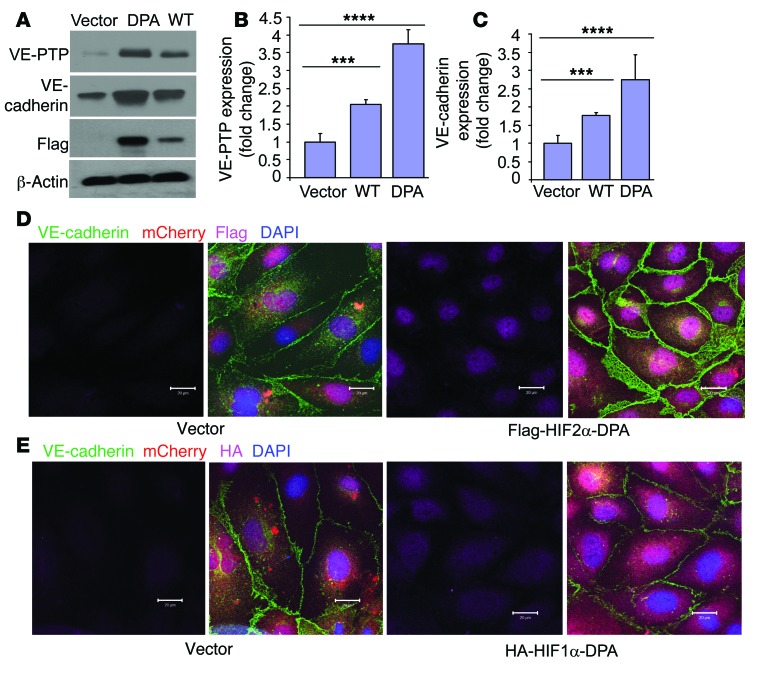
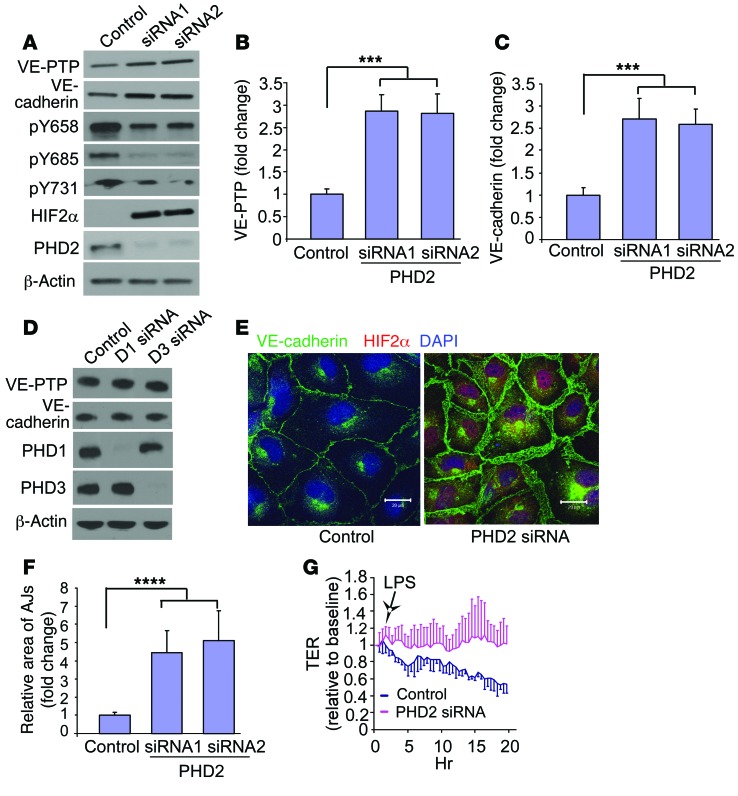
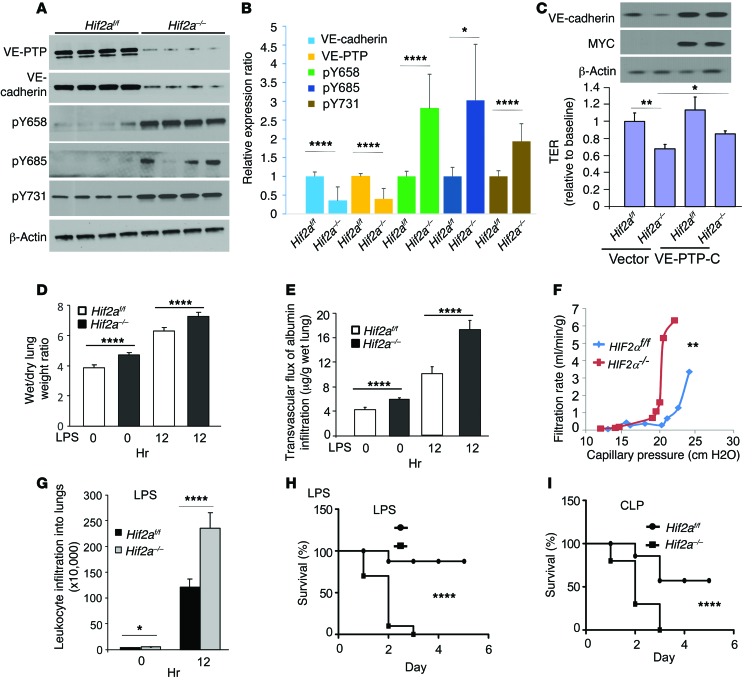
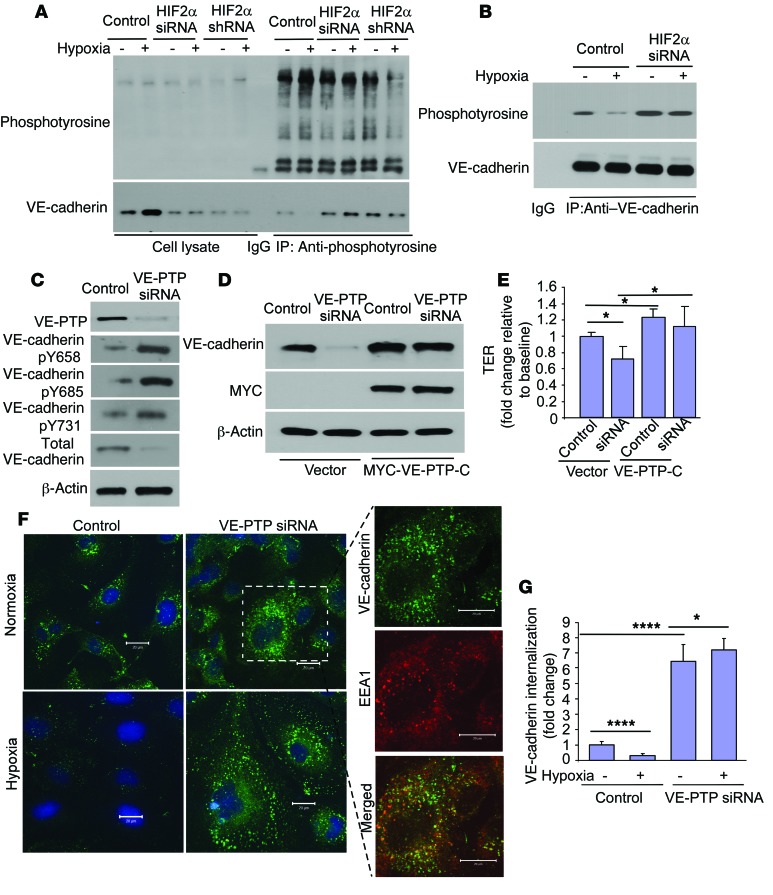
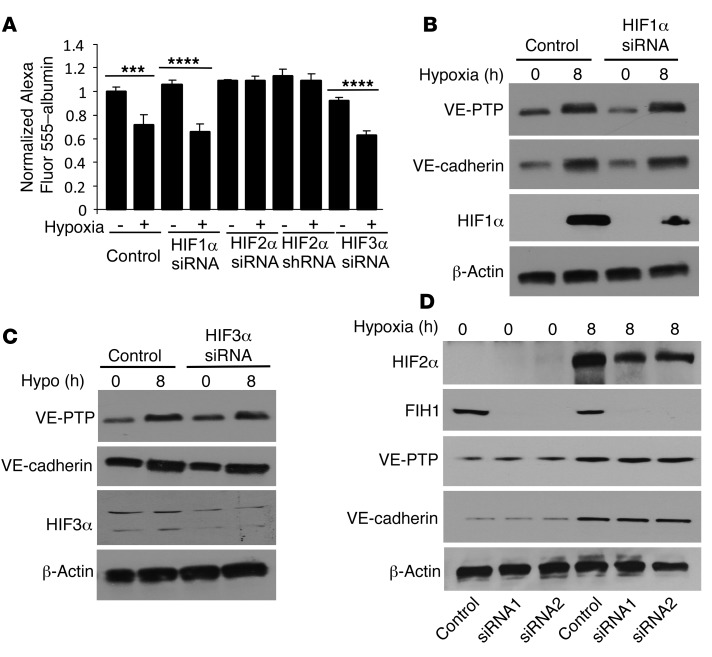
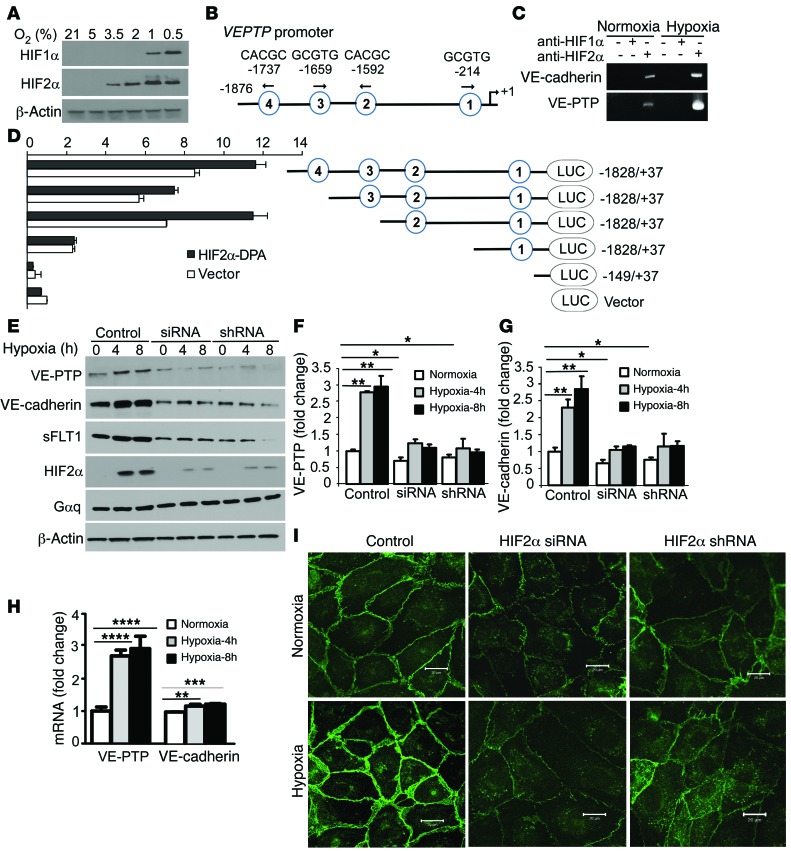
Vascular endothelial barrier dysfunction underlies diseases such as acute respiratory distress syndrome (ARDS), characterized by edema and inflammatory cell infiltration. The transcription factor HIF2α is highly expressed in vascular endothelial cells (ECs) and may regulate endothelial barrier function. Here, we analyzed promoter sequences of genes encoding proteins that regulate adherens junction (AJ) integrity and determined that vascular endothelial protein tyrosine phosphatase (VE-PTP) is a HIF2α target. HIF2α-induced VE-PTP expression enhanced dephosphorylation of VE-cadherin, which reduced VE-cadherin endocytosis and thereby augmented AJ integrity and endothelial barrier function. Mice harboring an EC-specific deletion of Hif2a exhibited decreased VE-PTP expression and increased VE-cadherin phosphorylation, resulting in defective AJs. Mice lacking HIF2α in ECs had increased lung vascular permeability and water content, both of which were further exacerbated by endotoxin-mediated injury. Treatment of these mice with Fg4497, a prolyl hydroxylase domain 2 (PHD2) inhibitor, activated HIF2α-mediated transcription in a hypoxia-independent manner. HIF2α activation increased VE-PTP expression, decreased VE-cadherin phosphorylation, promoted AJ integrity, and prevented the loss of endothelial barrier function. These findings demonstrate that HIF2α enhances endothelial barrier integrity, in part through VE-PTP expression and the resultant VE-cadherin dephosphorylation-mediated assembly of AJs. Moreover, activation of HIF2α/VE-PTP signaling via PHD2 inhibition has the potential to prevent the formation of leaky vessels and edema in inflammatory diseases such as ARDS.
Figures
References
-
- Farahani RM, Sarrafpour B, Simonian M, Li Q, Hunter N. Directed glia-assisted angiogenesis in a mature neurosensory structure: pericytes mediate an adaptive response in human dental pulp that maintains blood-barrier function. J Comp Neurol. 2012;520(17):3803–3826. doi: 10.1002/cne.23162. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
