

Global transcriptional disturbances underlie Cornelia de Lange syndrome and related phenotypes
- PMID: 25574841
- PMCID: PMC4319410
- DOI: 10.1172/JCI77435
Global transcriptional disturbances underlie Cornelia de Lange syndrome and related phenotypes
Abstract
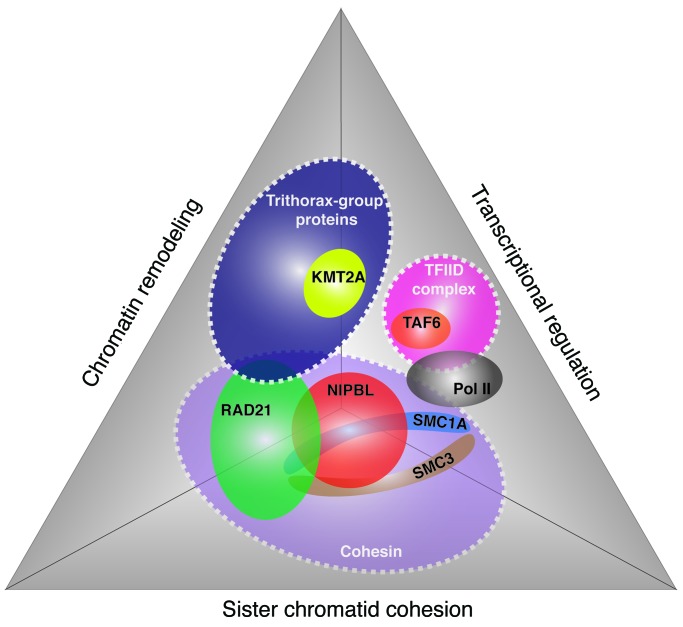
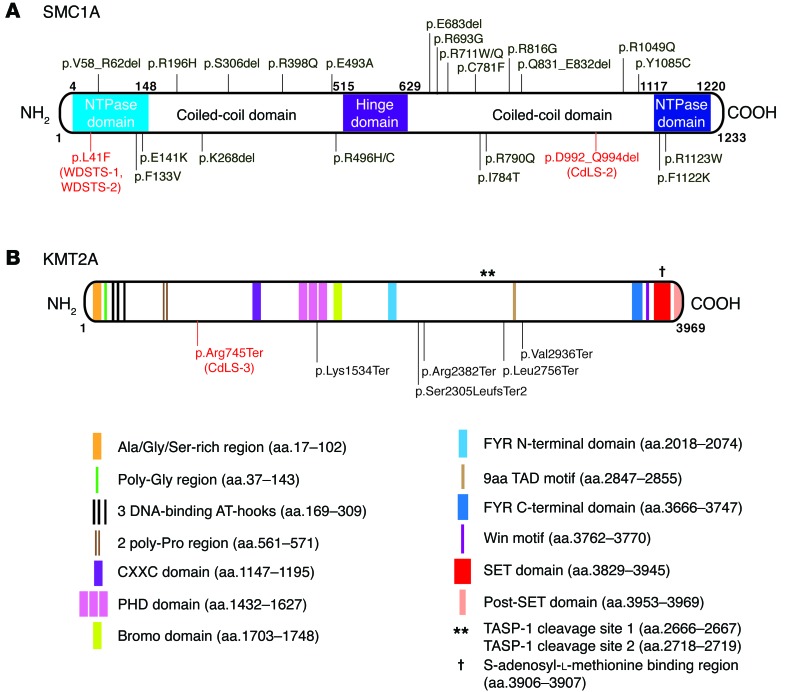
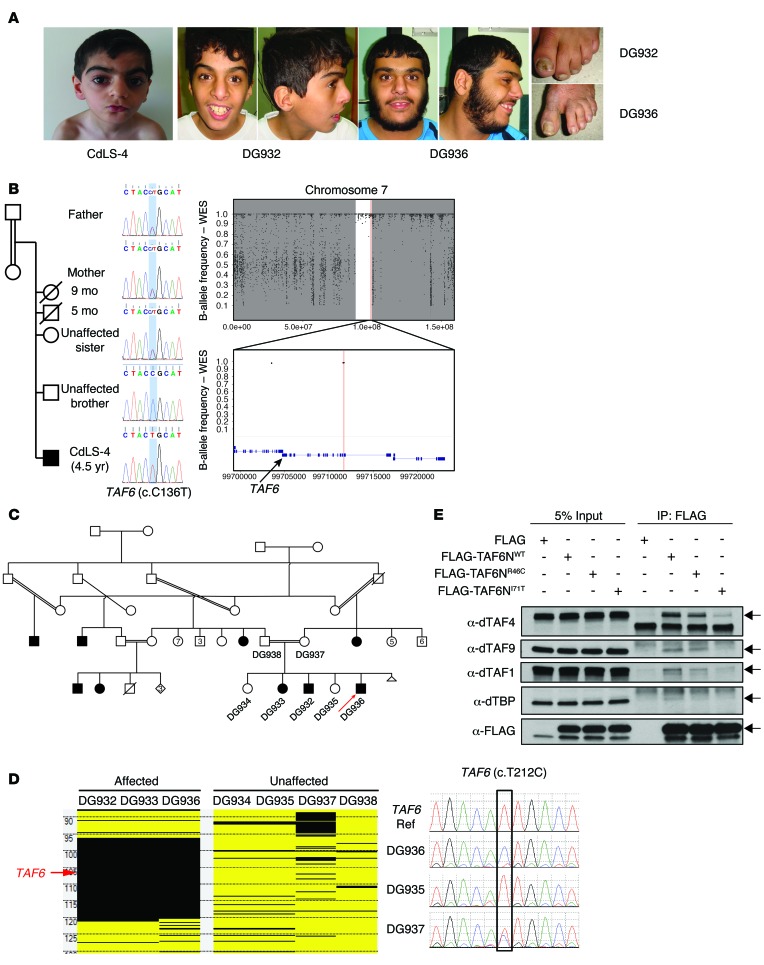
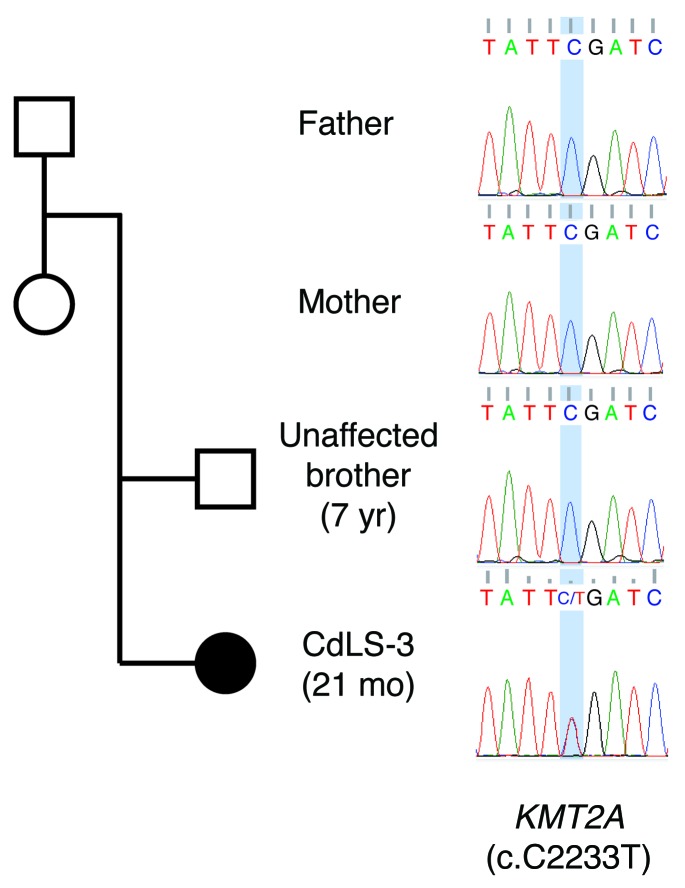
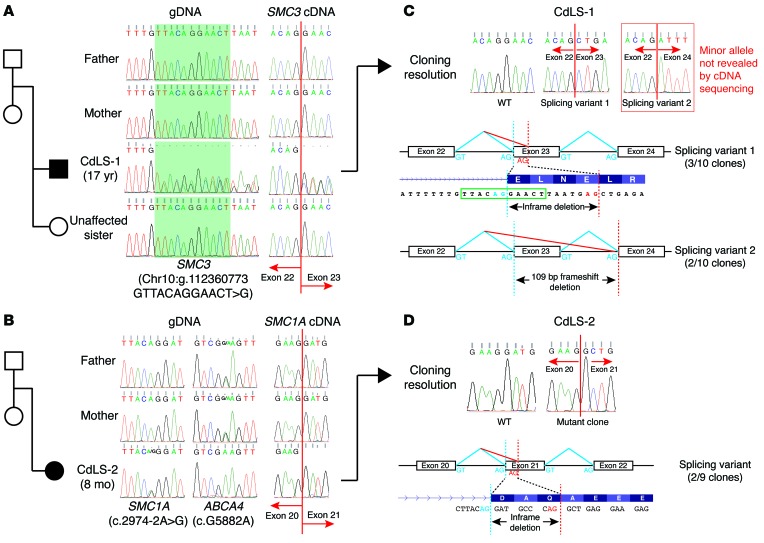
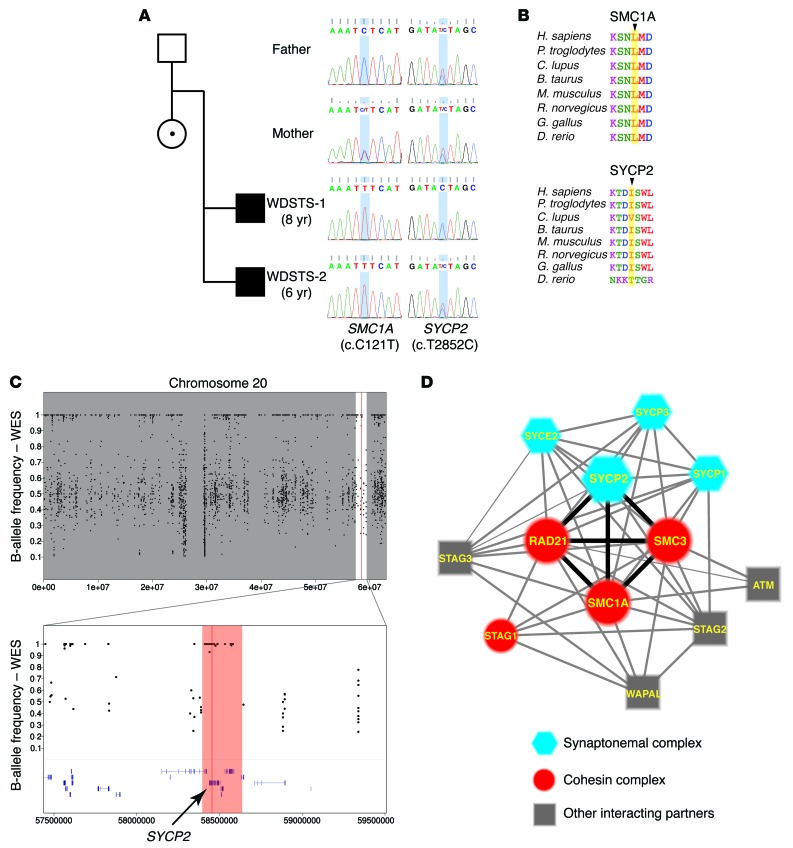
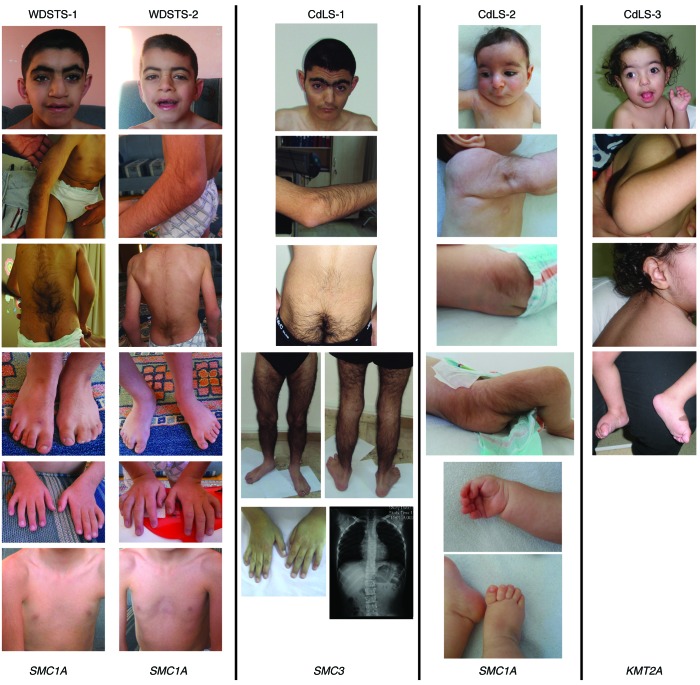
Cornelia de Lange syndrome (CdLS) is a genetically heterogeneous disorder that presents with extensive phenotypic variability, including facial dysmorphism, developmental delay/intellectual disability (DD/ID), abnormal extremities, and hirsutism. About 65% of patients harbor mutations in genes that encode subunits or regulators of the cohesin complex, including NIPBL, SMC1A, SMC3, RAD21, and HDAC8. Wiedemann-Steiner syndrome (WDSTS), which shares CdLS phenotypic features, is caused by mutations in lysine-specific methyltransferase 2A (KMT2A). Here, we performed whole-exome sequencing (WES) of 2 male siblings clinically diagnosed with WDSTS; this revealed a hemizygous, missense mutation in SMC1A that was predicted to be deleterious. Extensive clinical evaluation and WES of 32 Turkish patients clinically diagnosed with CdLS revealed the presence of a de novo heterozygous nonsense KMT2A mutation in 1 patient without characteristic WDSTS features. We also identified de novo heterozygous mutations in SMC3 or SMC1A that affected RNA splicing in 2 independent patients with combined CdLS and WDSTS features. Furthermore, in families from 2 separate world populations segregating an autosomal-recessive disorder with CdLS-like features, we identified homozygous mutations in TAF6, which encodes a core transcriptional regulatory pathway component. Together, our data, along with recent transcriptome studies, suggest that CdLS and related phenotypes may be "transcriptomopathies" rather than cohesinopathies.
Figures
References
-
- Brachmann W. Ein fall von symmetrischer monodaktylie durch Ulnadefekt, mit symmetrischer flughautbildung in den ellenbeugen, sowie anderen abnormitaten (zwerghaftogkeit, halsrippen, behaarung). Jarb Kinder Phys Erzie. 1916;84:225–235.
-
- de Lange C. Sur un type nouveau de degenerescence (typus Amstelodamensis). Arch Med Enfants. 1933;36:713–719.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
