

Amyloid deposits and inflammatory infiltrates in sporadic inclusion body myositis: the inflammatory egg comes before the degenerative chicken
- PMID: 25579751
- PMCID: PMC4405277
- DOI: 10.1007/s00401-015-1384-5
Amyloid deposits and inflammatory infiltrates in sporadic inclusion body myositis: the inflammatory egg comes before the degenerative chicken
Abstract
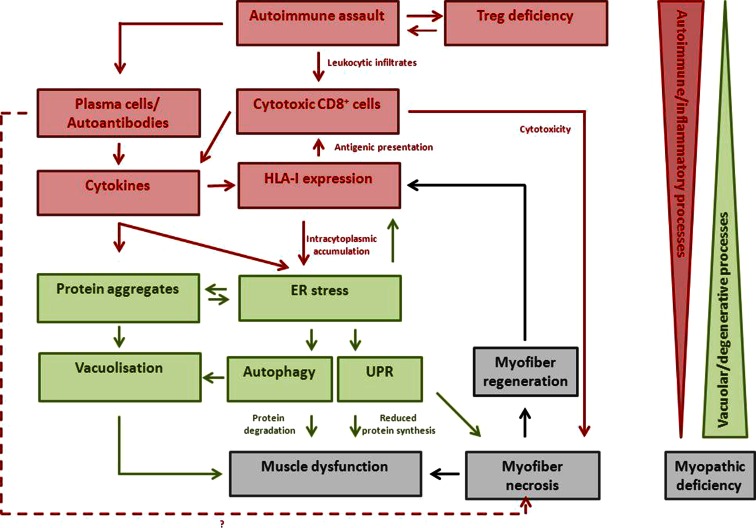
Sporadic inclusion body myositis (sIBM) is the most frequently acquired myopathy in patients over 50 years of age. It is imperative that neurologists and rheumatologists recognize this disorder which may, through clinical and pathological similarities, mimic other myopathies, especially polymyositis. Whereas polymyositis responds to immunosuppressant drug therapy, sIBM responds poorly, if at all. Controversy reigns as to whether sIBM is primarily an inflammatory or a degenerative myopathy, the distinction being vitally important in terms of directing research for effective specific therapies. We review here the pros and the cons for the respective hypotheses. A possible scenario, which our experience leads us to favour, is that sIBM may start with inflammation within muscle. The rush of leukocytes attracted by chemokines and cytokines may induce fibre injury and HLA-I overexpression. If the protein degradation systems are overloaded (possibly due to genetic predisposition, particular HLA-I subtypes or ageing), amyloid and other protein deposits may appear within muscle fibres, reinforcing the myopathic process in a vicious circle.
Figures
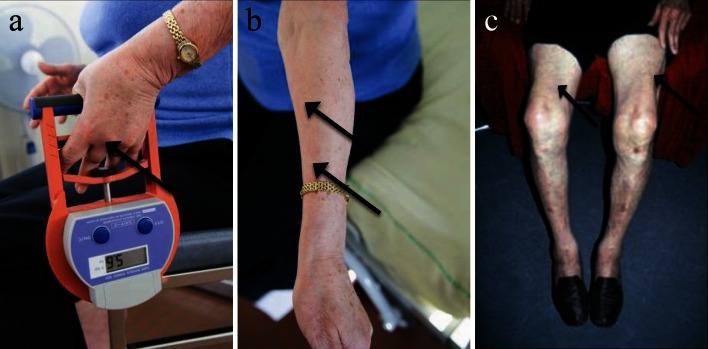
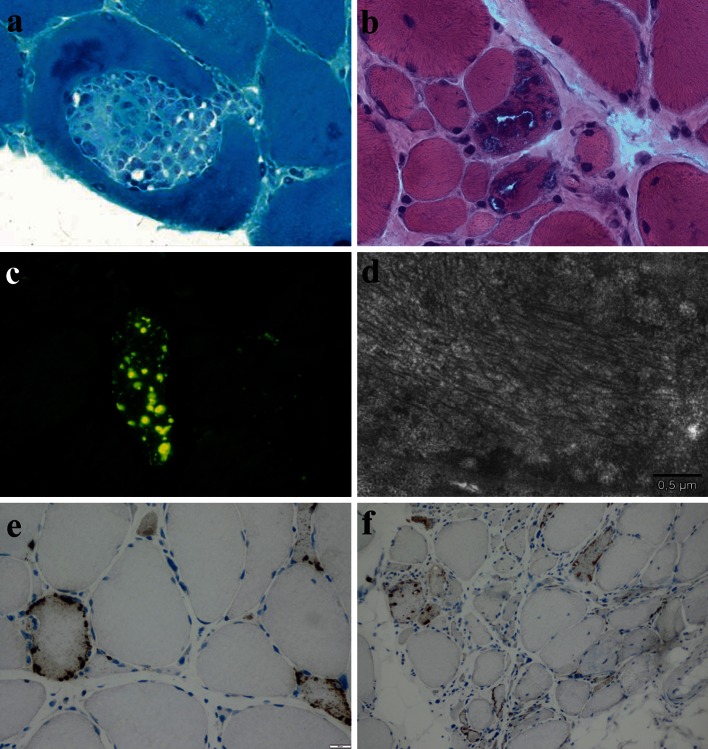
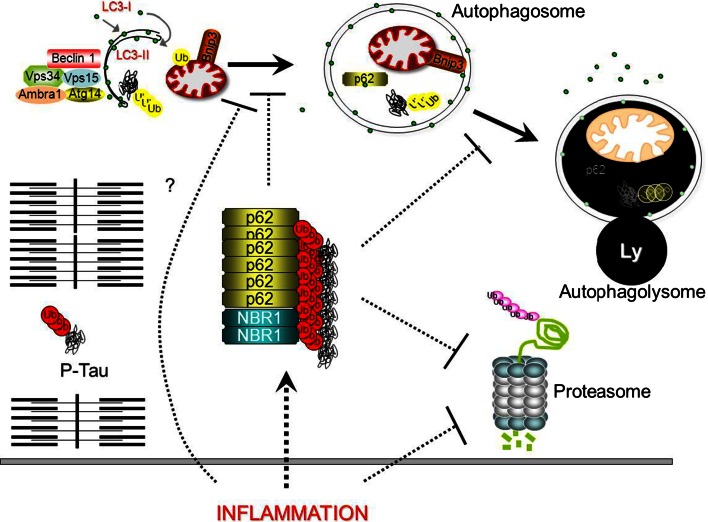
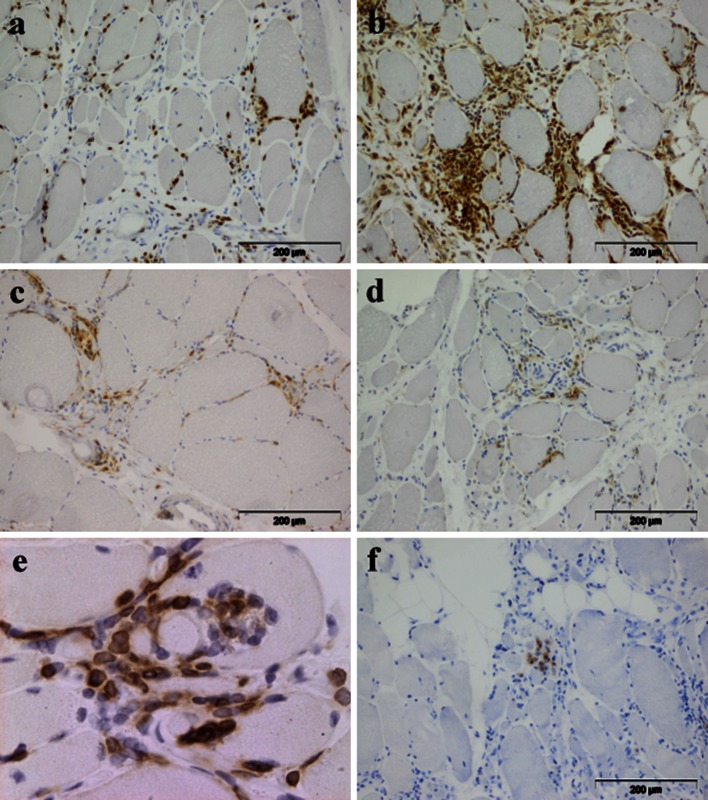
References
-
- Amato AA, Griggs RC. Unicorns, dragons, polymyositis, and other mythological beasts. Neurology. 2003;61(3):288–289. - PubMed
-
- Amemiya K, Granger RP, Dalakas MC. Clonal restriction of T-cell receptor expression by infiltrating lymphocytes in inclusion body myositis persists over time. Studies in repeated muscle biopsies. Brain. 2000;123(Pt 10):2030–2039. - PubMed
-
- Amouri R, Driss A, Murayama K, Kefi M, Nishino I, Hentati F. Allelic heterogeneity of GNE gene mutation in two Tunisian families with autosomal recessive inclusion body myopathy. Neuromuscul Disord NMD. 2005;15(5):361–363. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
