

Complex host genetics influence the microbiome in inflammatory bowel disease
- PMID: 25587358
- PMCID: PMC4292994
- DOI: 10.1186/s13073-014-0107-1
Complex host genetics influence the microbiome in inflammatory bowel disease
Abstract
Background: Human genetics and host-associated microbial communities have been associated independently with a wide range of chronic diseases. One of the strongest associations in each case is inflammatory bowel disease (IBD), but disease risk cannot be explained fully by either factor individually. Recent findings point to interactions between host genetics and microbial exposures as important contributors to disease risk in IBD. These include evidence of the partial heritability of the gut microbiota and the conferral of gut mucosal inflammation by microbiome transplant even when the dysbiosis was initially genetically derived. Although there have been several tests for association of individual genetic loci with bacterial taxa, there has been no direct comparison of complex genome-microbiome associations in large cohorts of patients with an immunity-related disease.
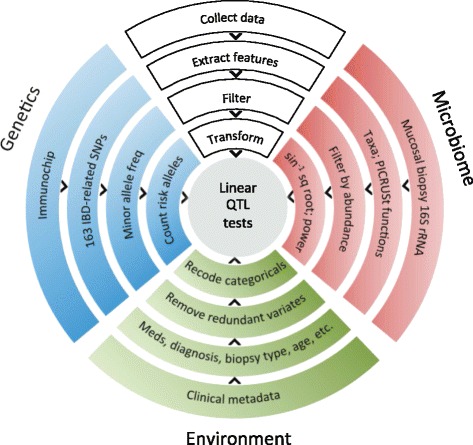
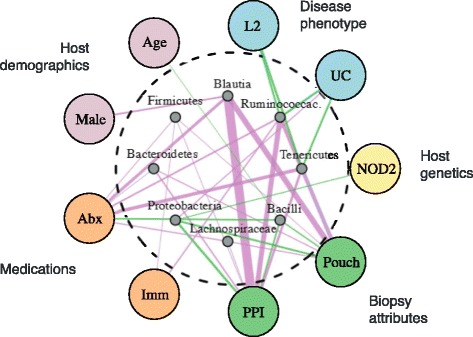
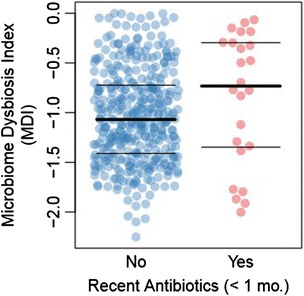
Methods: We obtained 16S ribosomal RNA (rRNA) gene sequences from intestinal biopsies as well as host genotype via Immunochip in three independent cohorts totaling 474 individuals. We tested for correlation between relative abundance of bacterial taxa and number of minor alleles at known IBD risk loci, including fine mapping of multiple risk alleles in the Nucleotide-binding oligomerization domain-containing protein 2 (NOD2) gene exon. We identified host polymorphisms whose associations with bacterial taxa were conserved across two or more cohorts, and we tested related genes for enrichment of host functional pathways.
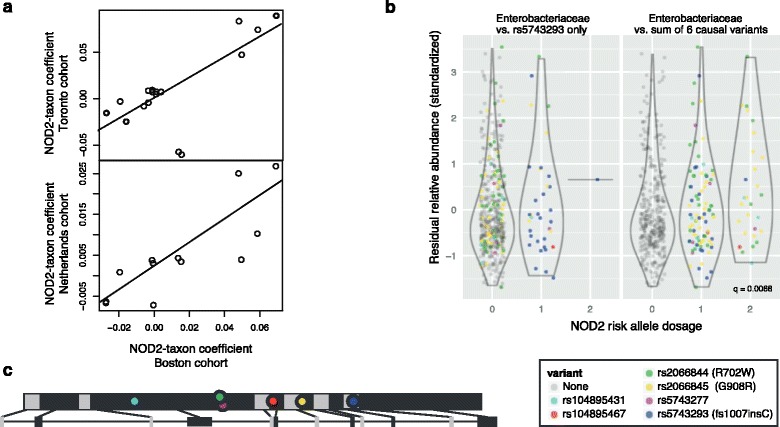
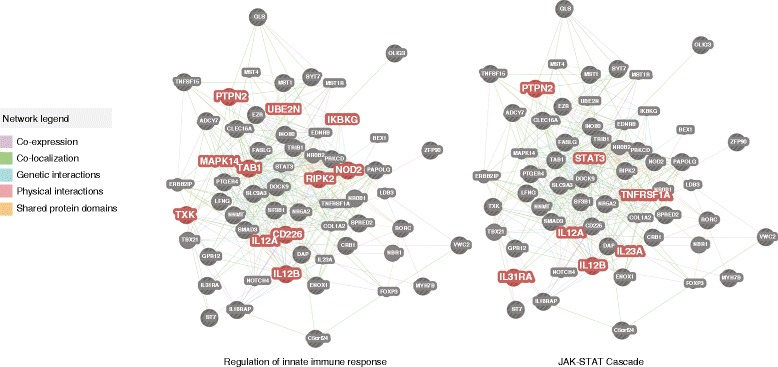
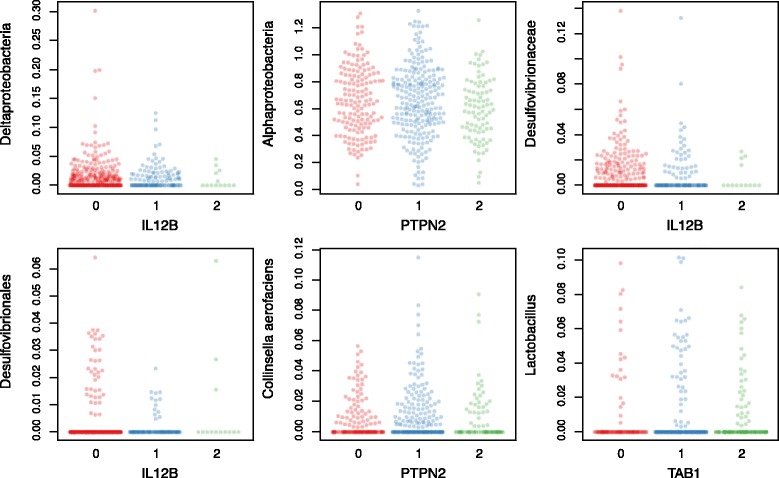
Results: We identified and confirmed in two cohorts a significant association between NOD2 risk allele count and increased relative abundance of Enterobacteriaceae, with directionality of the effect conserved in the third cohort. Forty-eight additional IBD-related SNPs have directionality of their associations with bacterial taxa significantly conserved across two or three cohorts, implicating genes enriched for regulation of innate immune response, the JAK-STAT cascade, and other immunity-related pathways.
Conclusions: These results suggest complex interactions between genetically altered host functional pathways and the structure of the microbiome. Our findings demonstrate the ability to uncover novel associations from paired genome-microbiome data, and they suggest a complex link between host genetics and microbial dysbiosis in subjects with IBD across independent cohorts.
Figures
References
-
- Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, Essers J, Mitrovic M, Ning K, Cleynen I, Theatre E, Spain SL, Raychaudhuri S, Goyette P, Wei Z, Abraham C, Achkar JP, Ahmad T, Amininejad L, Ananthakrishnan AN, Andersen V, Andrews JM, Baidoo L, Balschun T, Bampton PA, Bitton A, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. doi: 10.1038/nature11582. - DOI - PMC - PubMed
-
- Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV, Reyes JA, Shah SA, LeLeiko N, Snapper SB, Bousvaros A, Korzenik J, Sands BE, Xavier RJ, Huttenhower C. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012;13:R79. doi: 10.1186/gb-2012-13-9-r79. - DOI - PMC - PubMed
-
- Baumgart M, Dogan B, Rishniw M, Weitzman G, Bosworth B, Yantiss R, Orsi RH, Wiedmann M, McDonough P, Kim SG, Berg D, Schukken Y, Scherl E, Simpson KW. Culture independent analysis of ileal mucosa reveals a selective increase in invasive Escherichia coli of novel phylogeny relative to depletion of Clostridiales in Crohn’s disease involving the ileum. ISME J. 2007;1:403–418. doi: 10.1038/ismej.2007.52. - DOI - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
