

CCR2 antagonism alters brain macrophage polarization and ameliorates cognitive dysfunction induced by traumatic brain injury
- PMID: 25589768
- PMCID: PMC4293420
- DOI: 10.1523/JNEUROSCI.2405-14.2015
CCR2 antagonism alters brain macrophage polarization and ameliorates cognitive dysfunction induced by traumatic brain injury
Abstract
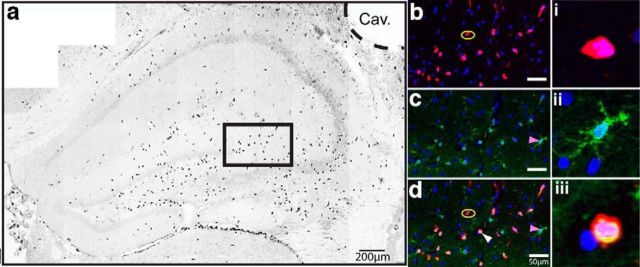
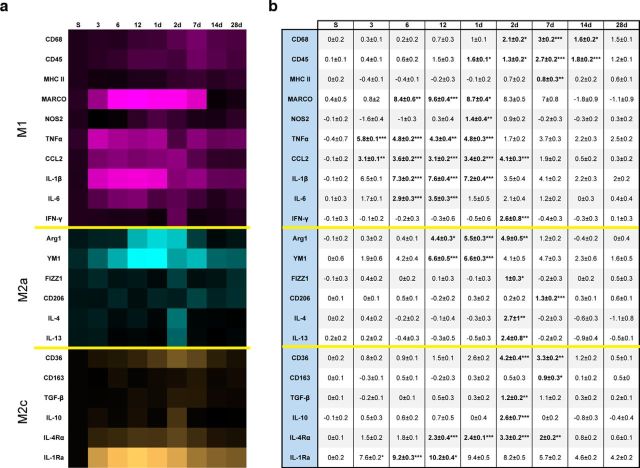
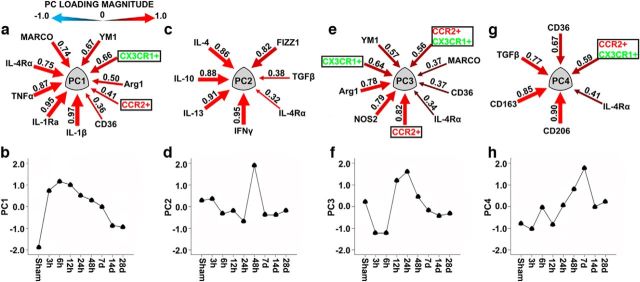
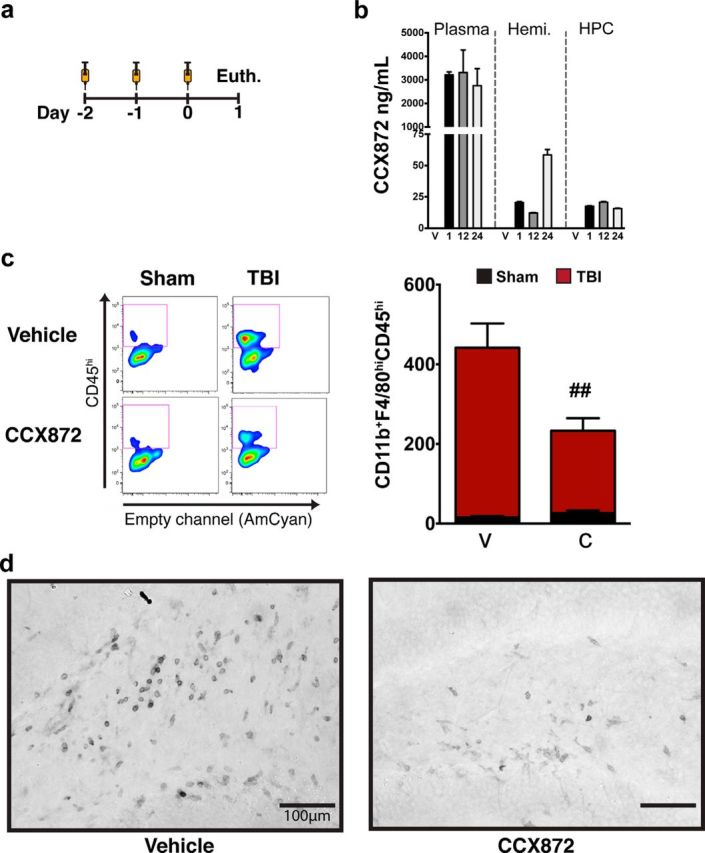
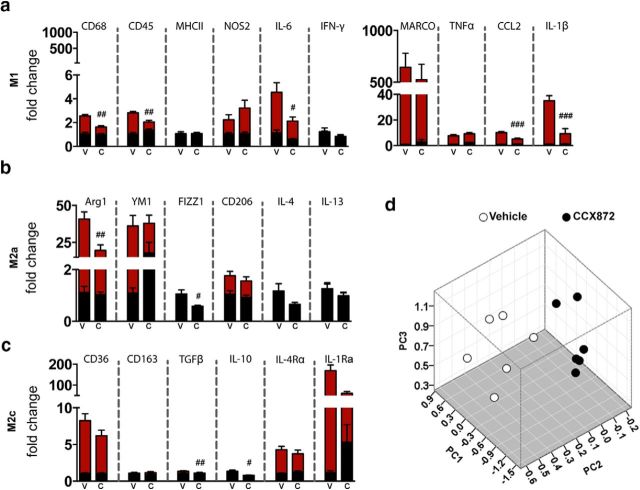
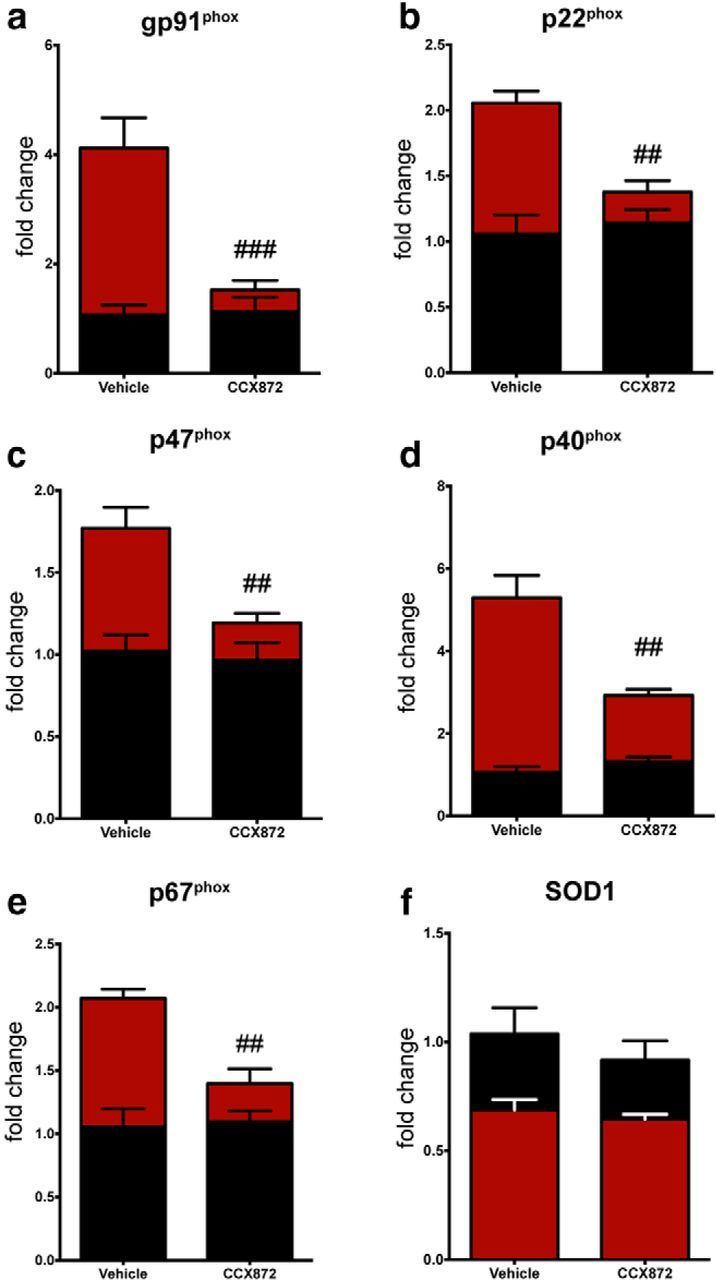
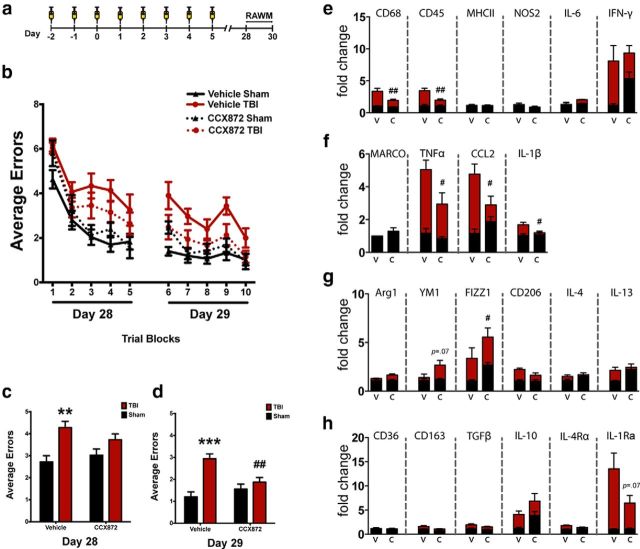
Traumatic brain injury (TBI) is a major risk factor for the development of multiple neurodegenerative diseases. With respect to the increasing prevalence of TBI, new therapeutic strategies are urgently needed that will prevent secondary damage to primarily unaffected tissue. Consistently, neuroinflammation has been implicated as a key mediator of secondary damage following the initial mechanical insult. Following injury, there is uncertainty regarding the role that accumulating CCR2(+) macrophages play in the injury-induced neuroinflammatory sequelae and cognitive dysfunction. Using CX3CR1(GFP/+)CCR2(RFP/+) reporter mice, we show that TBI initiated a temporally restricted accumulation of peripherally derived CCR2(+) macrophages, which were concentrated in the hippocampal formation, a region necessary for learning and memory. Multivariate analysis delineated CCR2(+) macrophages' neuroinflammatory response while identifying a novel therapeutic treatment window. As a proof of concept, targeting CCR2(+) macrophages with CCX872, a novel Phase I CCR2 selective antagonist, significantly reduced TBI-induced inflammatory macrophage accumulation. Concomitantly, there was a significant reduction in multiple proinflammatory and neurotoxic mediators with this treatment paradigm. Importantly, CCR2 antagonism resulted in a sparing of TBI-induced hippocampal-dependent cognitive dysfunction and reduced proinflammatory activation profile 1 month after injury. Thus, therapeutically targeting the CCR2(+) subset of monocytes/macrophages may provide a new avenue of clinical intervention following TBI.
Keywords: CCR2; PCA; TBI; inflammation; macrophage; therapeutic.
Copyright © 2015 the authors 0270-6474/15/350748-13$15.00/0.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials