

Divergent, coexisting Pseudomonas aeruginosa lineages in chronic cystic fibrosis lung infections
- PMID: 25590983
- PMCID: PMC4407486
- DOI: 10.1164/rccm.201409-1646OC
Divergent, coexisting Pseudomonas aeruginosa lineages in chronic cystic fibrosis lung infections
Abstract
Rationale: Pseudomonas aeruginosa, the predominant cause of chronic airway infections of patients with cystic fibrosis, exhibits extensive phenotypic diversity among isolates within and between sputum samples, but little is known about the underlying genetic diversity.
Objectives: To characterize the population genetic structure of transmissible P. aeruginosa Liverpool Epidemic Strain in chronic infections of nine patients with cystic fibrosis, and infer evolutionary processes associated with adaptation to the cystic fibrosis lung.
Methods: We performed whole-genome sequencing of P. aeruginosa isolates and pooled populations and used comparative analyses of genome sequences including phylogenetic reconstructions and resolution of population structure from genome-wide allele frequencies.
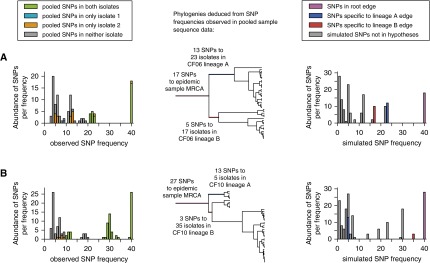
Measurements and main results: Genome sequences were obtained for 360 isolates from nine patients. Phylogenetic reconstruction of the ancestry of 40 individually sequenced isolates from one patient sputum sample revealed the coexistence of two genetically diverged, recombining lineages exchanging potentially adaptive mutations. Analysis of population samples for eight additional patients indicated coexisting lineages in six cases. Reconstruction of the ancestry of individually sequenced isolates from all patients indicated smaller genetic distances between than within patients in most cases.
Conclusions: Our population-level analysis demonstrates that coexistence of distinct lineages of P. aeruginosa Liverpool Epidemic Strain within individuals is common. In several cases, coexisting lineages may have been present in the infecting inoculum or assembled through multiple transmissions. Divergent lineages can share mutations via homologous recombination, potentially aiding adaptation to the airway during chronic infection. The genetic diversity of this transmissible strain within infections, revealed by high-resolution genomics, has implications for patient segregation and therapeutic strategies.
Keywords: bacteria; genomics; homologous recombination; population genetics.
Figures





Comment in
-
Show me the SNPs. How bacterial sex generates diversity in the cystic fibrosis lung.Am J Respir Crit Care Med. 2015 Apr 1;191(7):725-7. doi: 10.1164/rccm.201502-0248ED. Am J Respir Crit Care Med. 2015. PMID: 25830516 No abstract available.
References
-
- Driscoll JA, Brody SL, Kollef MH. The epidemiology, pathogenesis and treatment of Pseudomonas aeruginosa infections. Drugs. 2007;67:351–368. - PubMed
-
- Murray TS, Egan M, Kazmierczak BI. Pseudomonas aeruginosa chronic colonization in cystic fibrosis patients. Curr Opin Pediatr. 2007;19:83–88. - PubMed
-
- Goodman AL, Kulasekara B, Rietsch A, Boyd D, Smith RS, Lory S. A signaling network reciprocally regulates genes associated with acute infection and chronic persistence in Pseudomonas aeruginosa. Dev Cell. 2004;7:745–754. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
