

Demographic, clinical, and laboratory parameters of cystic fibrosis during the last two decades: a comparative analysis
- PMID: 25592785
- PMCID: PMC4417211
- DOI: 10.1186/1471-2466-15-3
Demographic, clinical, and laboratory parameters of cystic fibrosis during the last two decades: a comparative analysis
Abstract
Background: In recent years, patients with cystic fibrosis (CF) have tended to experience a longer life expectancy and higher quality of life. In this context, the aim of the present study was to evaluate and compare the demographic, clinical, and laboratory markers of patients with CF during the last two decades at a CF referral center.
Methods: A retrospective study of the demographic, clinical, and laboratory markers for CF treatment at a CF referral center was performed during two decades: 2000 (DI, 1990-2000, n = 104 patients) and 2010 (DII, 2000-2010, n = 181 patients).
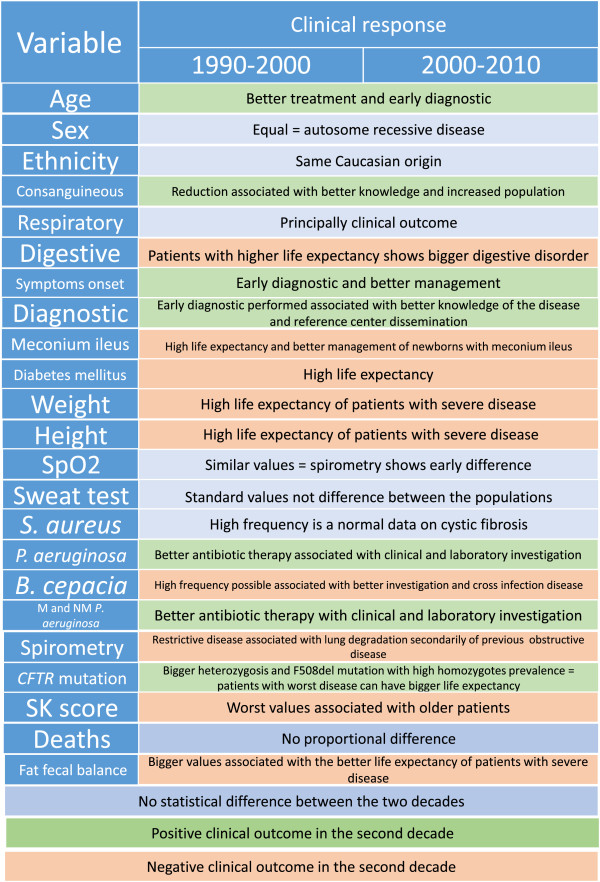
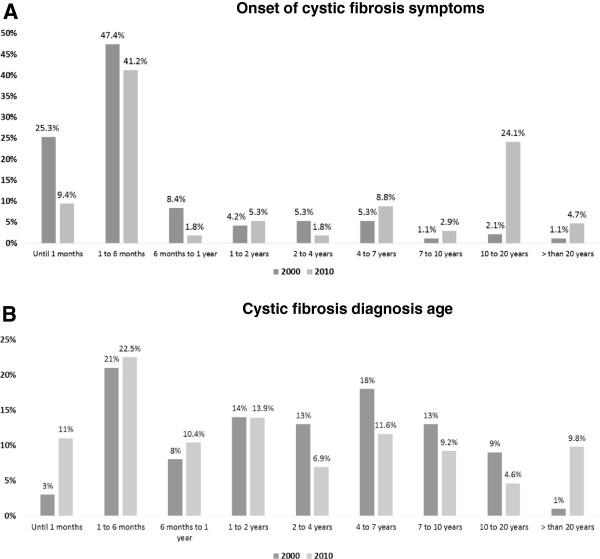
Results: The following variables were less common in DI than in DII: (i) pancreatic insufficiency, (ii) meconium ileus, (iii) diabetes mellitus, (iv) Burkholderia cepacia colonization, (v) moderate and severe Shwachman-Kulczycki score (SKS), (vi) F508del mutation screening, (vii) patients without an identified CFTR mutation (class IV, V, or VI mutation), (viii) patients above the 10th percentile for weight and height, (ix) restrictive lung disease, and (x) older patients (p < 0.01). The following variables were more common in DI than in DII: (i) excellent and good SKS, (ii) F508del heterozygous status, (iii) colonization by mucoid and nonmucoid Pseudomonas aeruginosa, (iv) obstructive lung disease, and (v) minimal time for CF diagnosis (p < 0.01).
Conclusion: Clinical outcomes differed between the two decades. Demographic, clinical, and laboratory markers in patients with CF are useful tools and should be encouraged in CF referral centers to determine the results of CF management and treatment, enabling a better understanding of this disease and its clinical evolution. Early diagnosis and management of CF will improve patients' quality of life and life expectancy until personalized drug therapy is possible for all patients with CF.
Figures
References
-
- Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL, Drumm ML, Iannuzzi MC, Collins FS, Tsui LC. Identification of the cystic fibrosis gene: cloning and characterization of the complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. - DOI - PubMed
-
- Rommens JM, Iannuzzi MC, Kerem BS, Drumm ML, Melmer G, Dean M, Rozmahel R, Cole JL, Kennedy D, Hidaka N, Zsiga M, Buchwald M, Riordan JR, Tsui LC, Collins FS. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989;245:1059–1065. doi: 10.1126/science.2772657. - DOI - PubMed
-
- Clinical and Functional Translation of CFTR. http://www.cftr2.org/
-
- NCBI Gene: CFTR cystic fibrosis transmembrane conductance regulator (ATP-binding cassette sub-family C, member 7) [Homo sapiens (human)] [http://www.ncbi.nlm.nih.gov/gene/1080]
Pre-publication history
-
- The pre-publication history for this paper can be accessed here: http://www.biomedcentral.com/1471-2466/15/3/prepub
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
