

Regulation of sarcoplasmic reticulum Ca2+ ATPase 2 (SERCA2) activity by phosphodiesterase 3A (PDE3A) in human myocardium: phosphorylation-dependent interaction of PDE3A1 with SERCA2
- PMID: 25593322
- PMCID: PMC4358103
- DOI: 10.1074/jbc.M115.638585
Regulation of sarcoplasmic reticulum Ca2+ ATPase 2 (SERCA2) activity by phosphodiesterase 3A (PDE3A) in human myocardium: phosphorylation-dependent interaction of PDE3A1 with SERCA2
Abstract
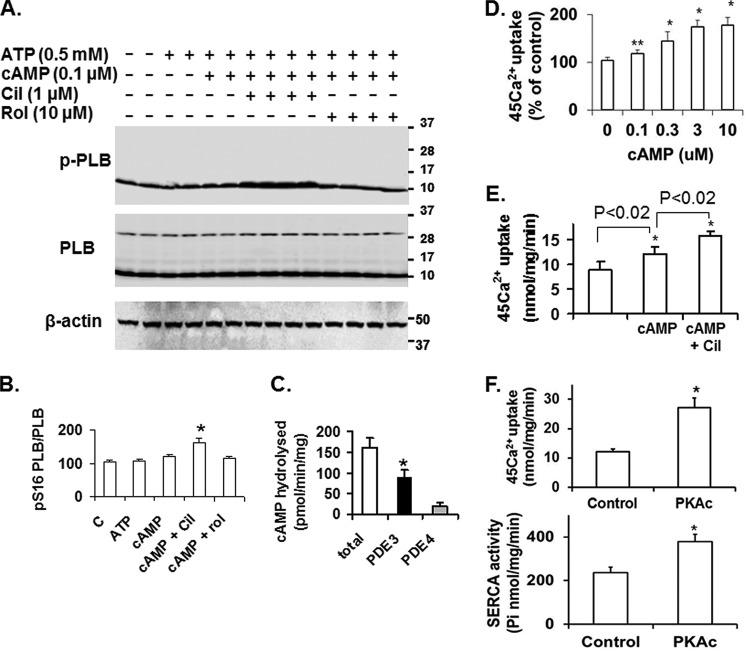
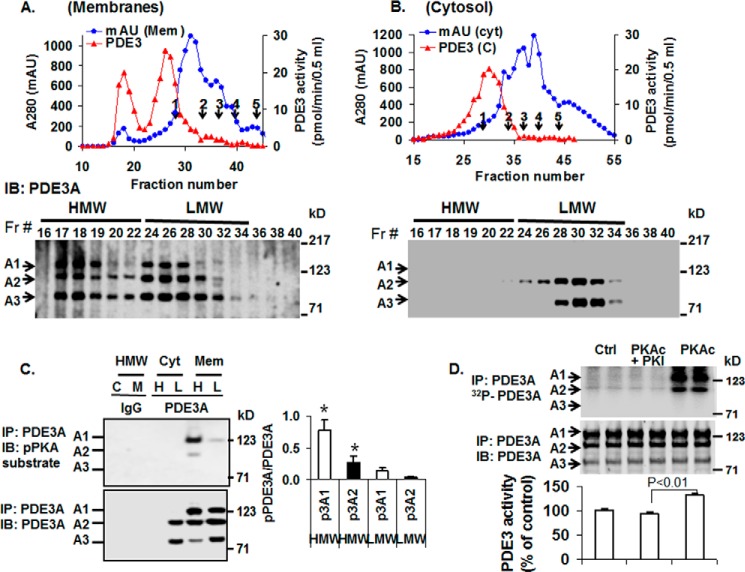
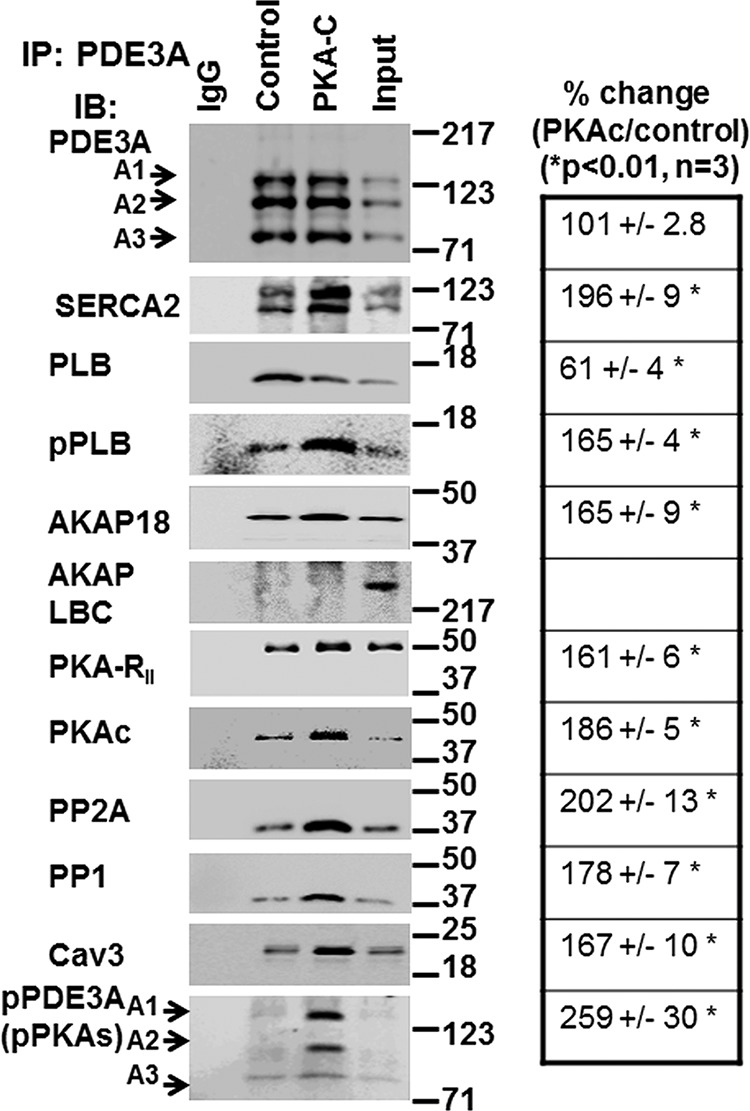
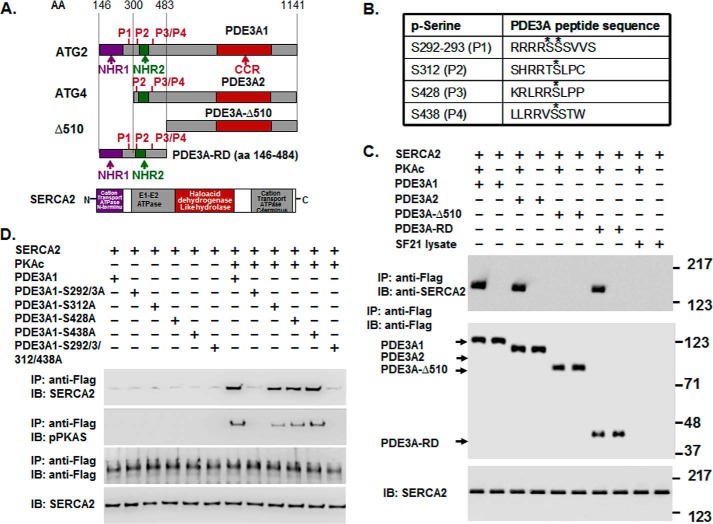
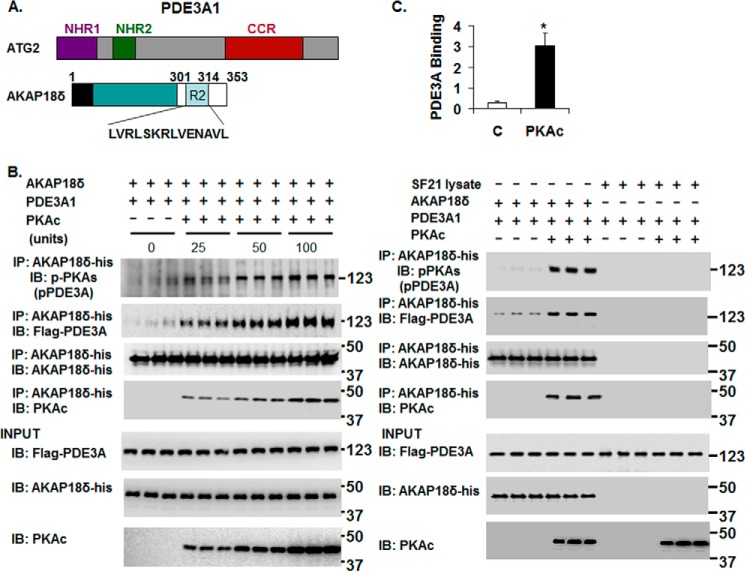
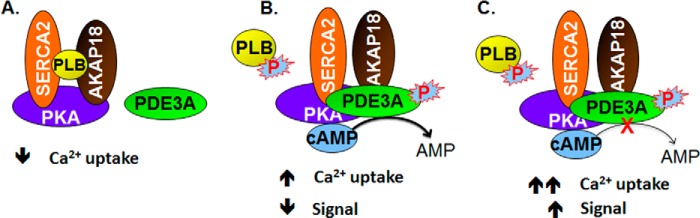
Cyclic nucleotide phosphodiesterase 3A (PDE3) regulates cAMP-mediated signaling in the heart, and PDE3 inhibitors augment contractility in patients with heart failure. Studies in mice showed that PDE3A, not PDE3B, is the subfamily responsible for these inotropic effects and that murine PDE3A1 associates with sarcoplasmic reticulum Ca(2+) ATPase 2 (SERCA2), phospholamban (PLB), and AKAP18 in a multiprotein signalosome in human sarcoplasmic reticulum (SR). Immunohistochemical staining demonstrated that PDE3A co-localizes in Z-bands of human cardiac myocytes with desmin, SERCA2, PLB, and AKAP18. In human SR fractions, cAMP increased PLB phosphorylation and SERCA2 activity; this was potentiated by PDE3 inhibition but not by PDE4 inhibition. During gel filtration chromatography of solubilized SR membranes, PDE3 activity was recovered in distinct high molecular weight (HMW) and low molecular weight (LMW) peaks. HMW peaks contained PDE3A1 and PDE3A2, whereas LMW peaks contained PDE3A1, PDE3A2, and PDE3A3. Western blotting showed that endogenous HMW PDE3A1 was the principal PKA-phosphorylated isoform. Phosphorylation of endogenous PDE3A by rPKAc increased cAMP-hydrolytic activity, correlated with shift of PDE3A from LMW to HMW peaks, and increased co-immunoprecipitation of SERCA2, cav3, PKA regulatory subunit (PKARII), PP2A, and AKAP18 with PDE3A. In experiments with recombinant proteins, phosphorylation of recombinant human PDE3A isoforms by recombinant PKA catalytic subunit increased co-immunoprecipitation with rSERCA2 and rat rAKAP18 (recombinant AKAP18). Deletion of the recombinant human PDE3A1/PDE3A2 N terminus blocked interactions with recombinant SERCA2. Serine-to-alanine substitutions identified Ser-292/Ser-293, a site unique to human PDE3A1, as the principal site regulating its interaction with SERCA2. These results indicate that phosphorylation of human PDE3A1 at a PKA site in its unique N-terminal extension promotes its incorporation into SERCA2/AKAP18 signalosomes, where it regulates a discrete cAMP pool that controls contractility by modulating phosphorylation-dependent protein-protein interactions, PLB phosphorylation, and SERCA2 activity.
Keywords: A-kinase Anchoring Protein (AKAP); Cyclic AMP (cAMP); Cyclic Nucleotide Phosphodiesterase; Immunohistochemistry; PDE3A; Phospholamban; Protein Kinase A (PKA); SERCA2; Subcellular Fractionation.
© 2015 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures


References
-
- Sun B., Li H., Shakur Y., Hensley J., Hockman S., Kambayashi J., Manganiello V. C., Liu Y. (2007) Role of phosphodiesterase type 3A and 3B in regulating platelet and cardiac function using subtype-selective knockout mice. Cell. Signal. 19, 1765–1771 - PubMed
-
- Beca S., Ahmad F., Shen W., Liu J., Makary S., Polidovitch N., Sun J., Hockman S., Chung Y. W., Movsesian M., Murphy E., Manganiello V., Backx P. H. (2013) Phosphodiesterase type 3A regulates basal myocardial contractility through interacting with sarcoplasmic reticulum calcium ATPase type 2a signaling complexes in mouse heart. Circ. Res. 112, 289–297 - PMC - PubMed
-
- Stefan E., Wiesner B., Baillie G. S., Mollajew R., Henn V., Lorenz D., Furkert J., Santamaria K., Nedvetsky P., Hundsrucker C., Beyermann M., Krause E., Pohl P., Gall I., MacIntyre A. N., Bachmann S., Houslay M. D., Rosenthal W., Klussmann E. (2007) Compartmentalization of cAMP-dependent signaling by phosphodiesterase-4D is involved in the regulation of vasopressin-mediated water reabsorption in renal principal cells. J. Am. Soc. Nephrol. 18, 199–212 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
