

PAPSS2 deficiency causes androgen excess via impaired DHEA sulfation--in vitro and in vivo studies in a family harboring two novel PAPSS2 mutations
- PMID: 25594860
- PMCID: PMC4399300
- DOI: 10.1210/jc.2014-3556
PAPSS2 deficiency causes androgen excess via impaired DHEA sulfation--in vitro and in vivo studies in a family harboring two novel PAPSS2 mutations
Abstract
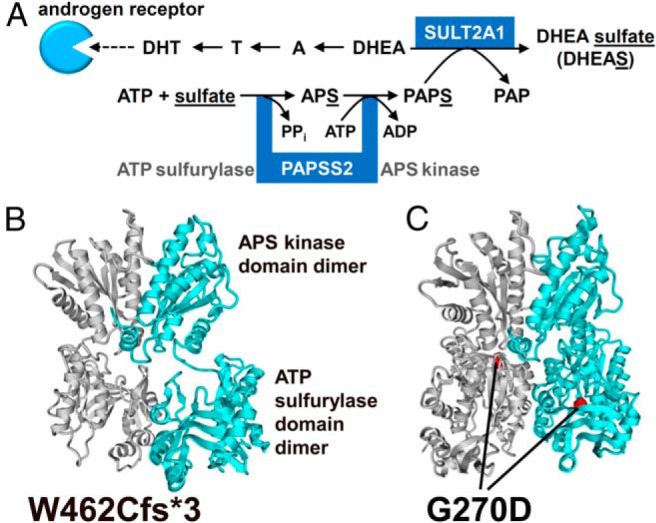
Context: PAPSS2 (PAPS synthase 2) provides the universal sulfate donor PAPS (3'-phospho-adenosine-5'-phosphosulfate) to all human sulfotransferases, including SULT2A1, responsible for sulfation of the crucial androgen precursor dehydroepiandrosterone (DHEA). Impaired DHEA sulfation is thought to increase the conversion of DHEA toward active androgens, a proposition supported by the previous report of a girl with inactivating PAPSS2 mutations who presented with low serum DHEA sulfate and androgen excess, clinically manifesting with premature pubarche and early-onset polycystic ovary syndrome.
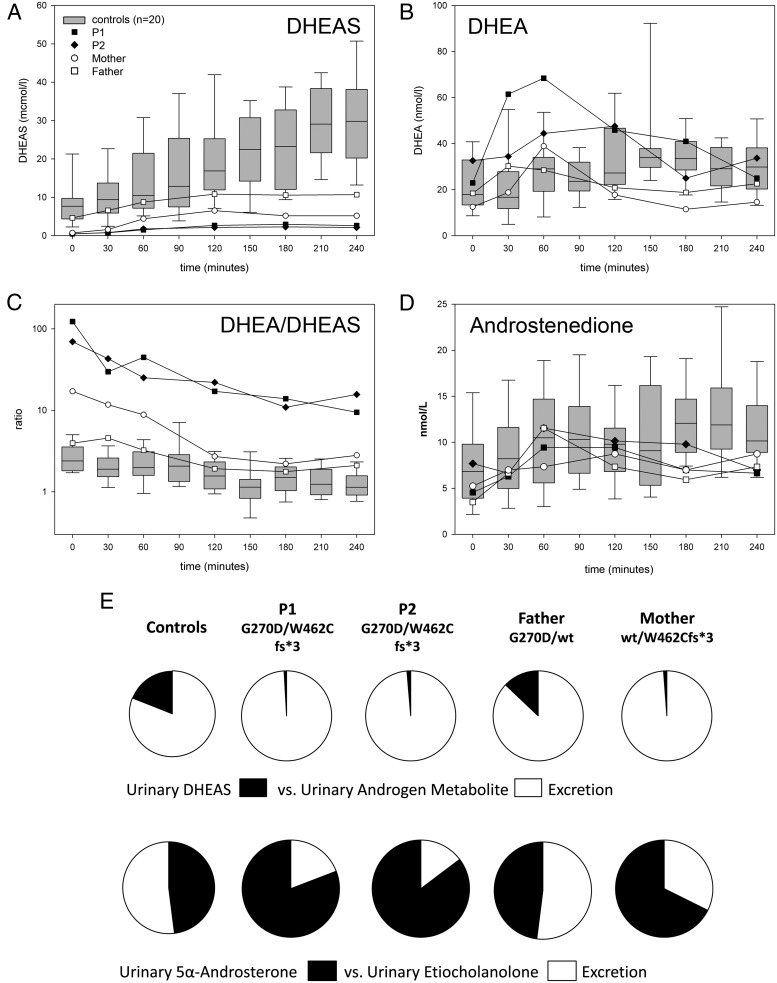
Patients and methods: We investigated a family harboring two novel PAPSS2 mutations, including two compound heterozygous brothers presenting with disproportionate short stature, low serum DHEA sulfate, but normal serum androgens. Patients and parents underwent a DHEA challenge test comprising frequent blood sampling and urine collection before and after 100 mg DHEA orally, with subsequent analysis of DHEA sulfation and androgen metabolism by mass spectrometry. The functional impact of the mutations was investigated in silico and in vitro.
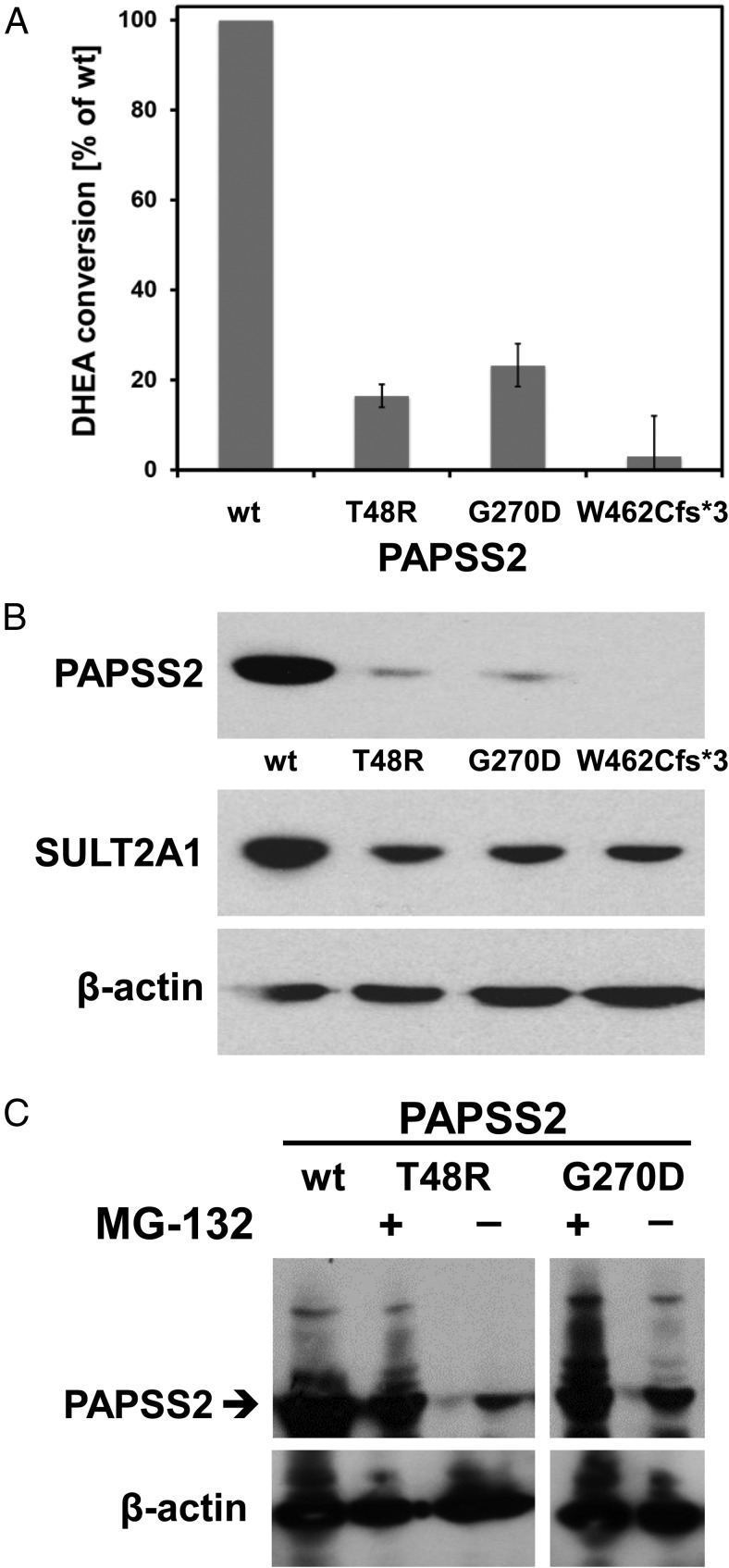
Results: We identified a novel PAPSS2 frameshift mutation, c.1371del, p.W462Cfs*3, resulting in complete disruption, and a novel missense mutation, c.809G>A, p.G270D, causing partial disruption of DHEA sulfation. Both patients and their mother, who was heterozygous for p.W462Cfs*3, showed increased 5α-reductase activity at baseline and significantly increased production of active androgens after DHEA intake. The mother had a history of oligomenorrhea and chronic anovulation that required clomiphene for ovulation induction.
Conclusions: We provide direct in vivo evidence for the significant functional impact of mutant PAPSS2 on DHEA sulfation and androgen activation. Heterozygosity for PAPSS2 mutations can be associated with a phenotype resembling polycystic ovary syndrome.
Figures
References
-
- Hammer F, Subtil S, Lux P, et al. No evidence for hepatic conversion of dehydroepiandrosterone (DHEA) sulfate to DHEA: in vivo and in vitro studies. J Clin Endocrinol Metab. 2005;90:3600–3605. - PubMed
-
- Arlt W, Hammer F, Sanning P, et al. Dissociation of serum dehydroepiandrosterone and dehydroepiandrosterone sulfate in septic shock. J Clin Endocrinol Metab. 2006;91:2548–2554. - PubMed
-
- Purohit A, Foster PA. Steroid sulfatase inhibitors for estrogen- and androgen-dependent cancers. J Endocrinol. 2012;212:99–110. - PubMed
-
- Noordam C, Dhir V, McNelis JC, et al. Inactivating PAPSS2 mutations in a patient with premature pubarche. N Engl J Med. 2009;360:2310–2318. - PubMed
-
- Faiyaz Ul Haque M, King LM, Krakow D, et al. Mutations in orthologous genes in human spondyloepimetaphyseal dysplasia and the brachymorphic mouse. Nat Genet. 1998;20:157–162. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
