

ERBB3-independent activation of the PI3K pathway in EGFR-mutant lung adenocarcinomas
- PMID: 25596284
- PMCID: PMC4400867
- DOI: 10.1158/0008-5472.CAN-13-1625
ERBB3-independent activation of the PI3K pathway in EGFR-mutant lung adenocarcinomas
Abstract
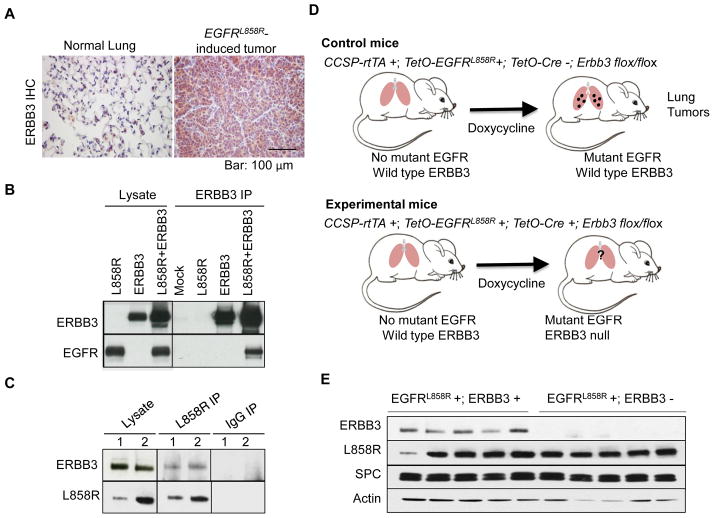
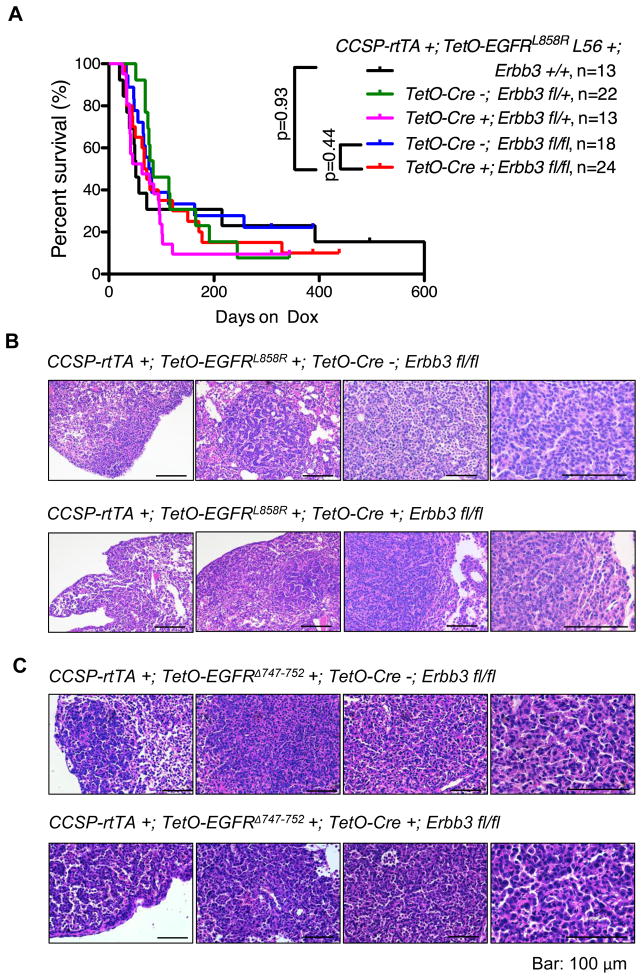
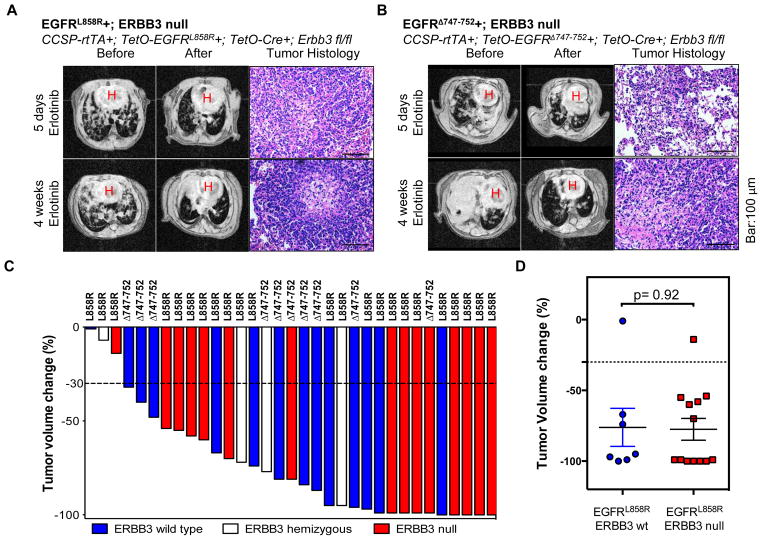
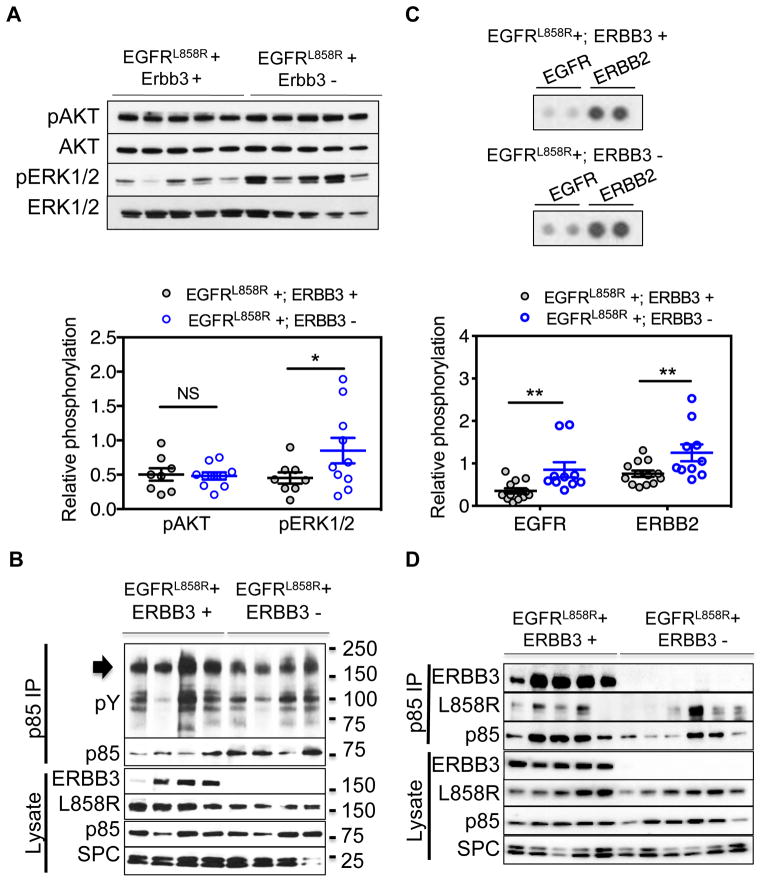
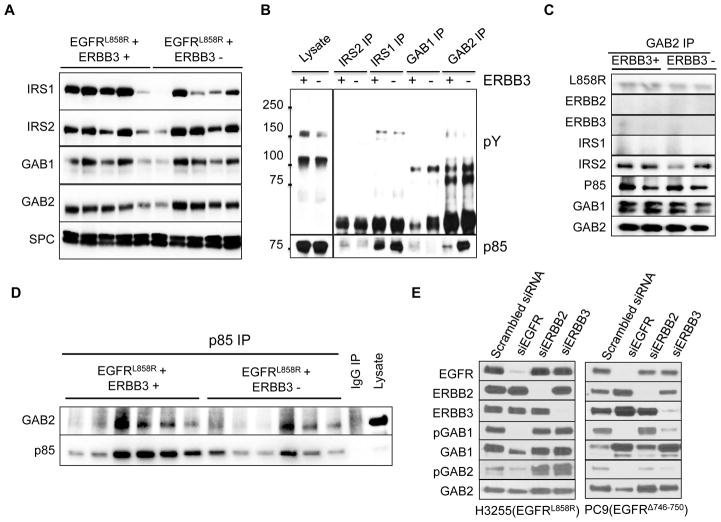
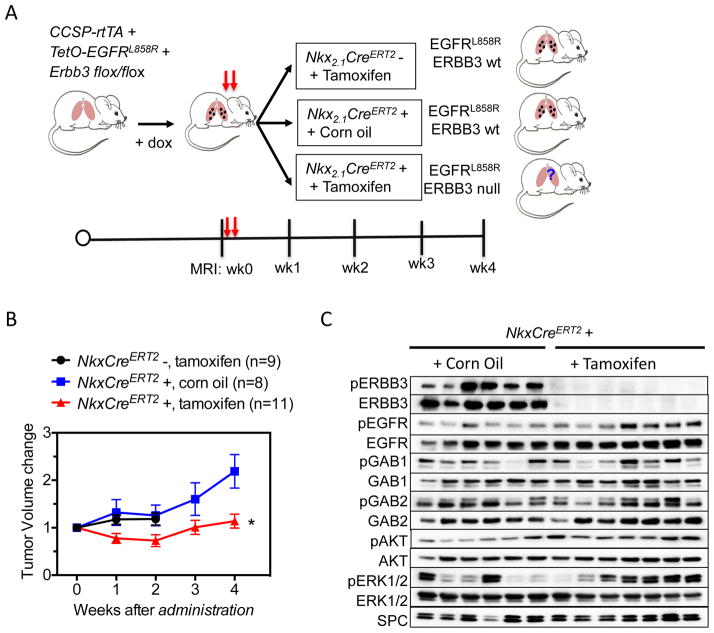
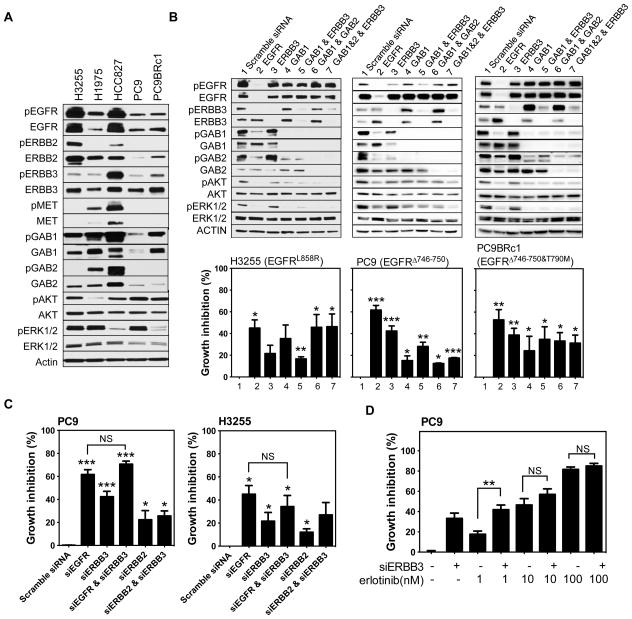
ERBB3, a member of the EGFR family of receptor tyrosine kinases, has been implicated in activation of the PI3K pathway in human lung adenocarcinomas driven by EGFR mutations. We investigated the contribution of ERBB3 to the initiation, progression, and therapeutic response of EGFR-induced lung adenocarcinomas using tetracycline- and tamoxifen-inducible transgenic mouse models. Deletion of Erbb3 at the time of induction of mutant EGFR had no effect on tumorigenesis, demonstrating that ERBB3 is not required to initiate tumorigenesis. Tumors that developed in the absence of ERBB3 remained sensitive to EGFR tyrosine kinase inhibitors and retained activation of the PI3K-AKT pathway. Interestingly, acute loss of Erbb3 suppressed further growth of established EGFR(L858R)-mediated lung tumors. Four weeks after deletion of Erbb3, the tumors exhibited phosphorylation of EGFR, of the adaptor proteins GAB1 and GAB2, and of the downstream signaling molecules AKT and ERK, suggesting that alternative signaling pathways could compensate for loss of Erbb3. Similar to our observations with mouse tumors, we found that GAB adaptor proteins play a role in ERBB3-independent activation of the PI3K pathway by mutant EGFR in EGFR-mutant human cell lines. Finally, in such cell lines, increased levels of phosphorylation of ERBB2 or MET were associated with reduced sensitivity to acute loss of ERBB3, suggesting remarkable plasticity in the signaling pathways regulated by mutant EGFR with important therapeutic implications.
©2015 American Association for Cancer Research.
Conflict of interest statement
Figures
References
-
- Riese DJ, 2nd, Stern DF. Specificity within the EGF family/ErbB receptor family signaling network. BioEssays : news and reviews in molecular, cellular and developmental biology. 1998;20(1):41–8. - PubMed
-
- Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. The New England journal of medicine. 2004;350(21):2129–39. - PubMed
-
- Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science (New York, NY. 2004;304(5676):1497–500. - PubMed
-
- Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(36):13306–11. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
