

CLPB mutations cause 3-methylglutaconic aciduria, progressive brain atrophy, intellectual disability, congenital neutropenia, cataracts, movement disorder
- PMID: 25597510
- PMCID: PMC4320260
- DOI: 10.1016/j.ajhg.2014.12.013
CLPB mutations cause 3-methylglutaconic aciduria, progressive brain atrophy, intellectual disability, congenital neutropenia, cataracts, movement disorder
Abstract
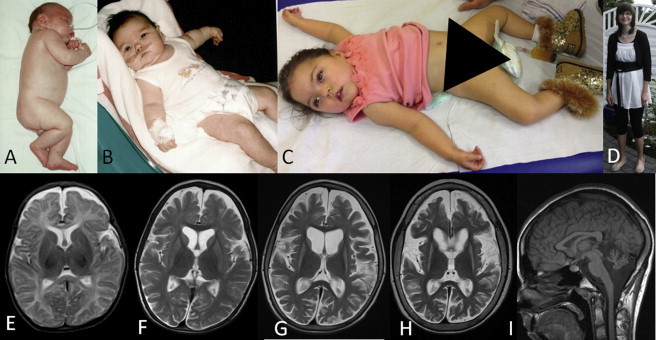
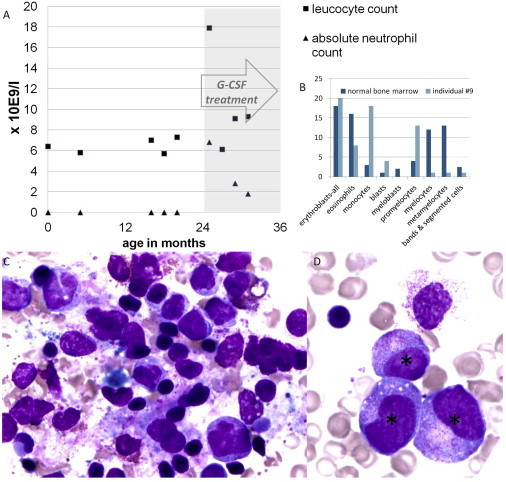
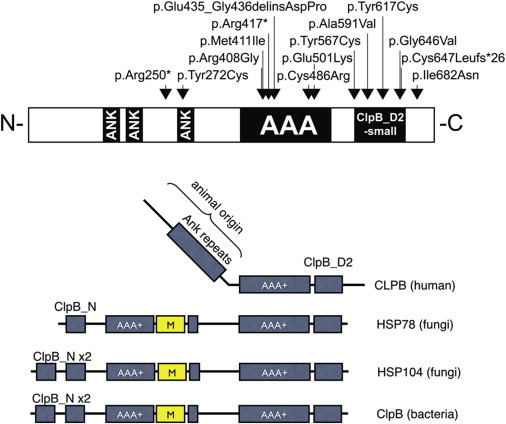
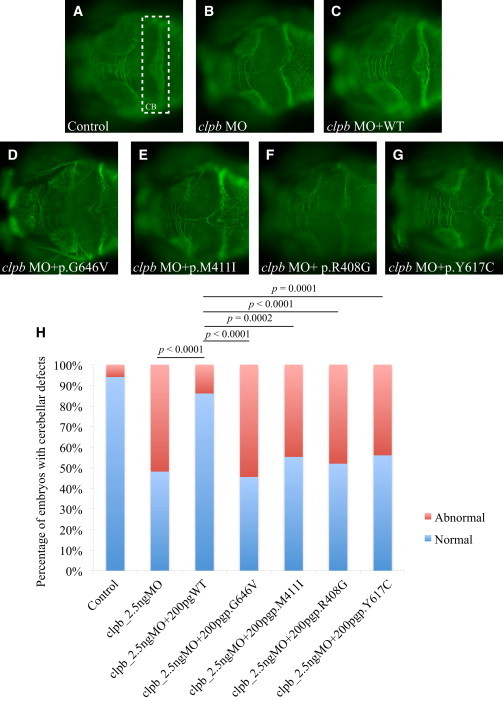
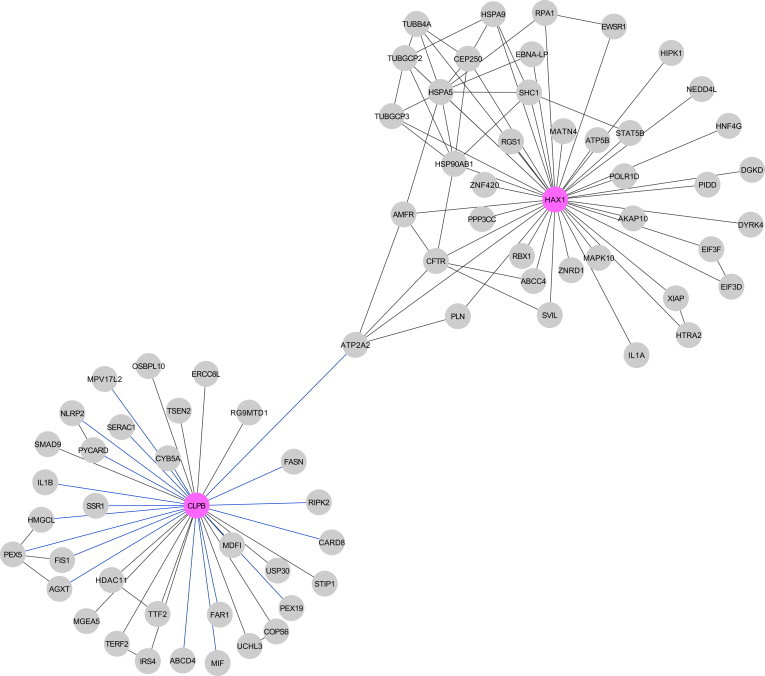
We studied a group of individuals with elevated urinary excretion of 3-methylglutaconic acid, neutropenia that can develop into leukemia, a neurological phenotype ranging from nonprogressive intellectual disability to a prenatal encephalopathy with progressive brain atrophy, movement disorder, cataracts, and early death. Exome sequencing of two unrelated individuals and subsequent Sanger sequencing of 16 individuals with an overlapping phenotype identified a total of 14 rare, predicted deleterious alleles in CLPB in 14 individuals from 9 unrelated families. CLPB encodes caseinolytic peptidase B homolog ClpB, a member of the AAA+ protein family. To evaluate the relevance of CLPB in the pathogenesis of this syndrome, we developed a zebrafish model and an in vitro assay to measure ATPase activity. Suppression of clpb in zebrafish embryos induced a central nervous system phenotype that was consistent with cerebellar and cerebral atrophy that could be rescued by wild-type, but not mutant, human CLPB mRNA. Consistent with these data, the loss-of-function effect of one of the identified variants (c.1222A>G [p.Arg408Gly]) was supported further by in vitro evidence with the mutant peptides abolishing ATPase function. Additionally, we show that CLPB interacts biochemically with ATP2A2, known to be involved in apoptotic processes in severe congenital neutropenia (SCN) 3 (Kostmann disease [caused by HAX1 mutations]). Taken together, mutations in CLPB define a syndrome with intellectual disability, congenital neutropenia, progressive brain atrophy, movement disorder, cataracts, and 3-methylglutaconic aciduria.
Copyright © 2015 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Wortmann S.B., Vaz F.M., Gardeitchik T., Vissers L.E., Renkema G.H., Schuurs-Hoeijmakers J.H., Kulik W., Lammens M., Christin C., Kluijtmans L.A. Mutations in the phospholipid remodeling gene SERAC1 impair mitochondrial function and intracellular cholesterol trafficking and cause dystonia and deafness. Nat. Genet. 2012;44:797–802. - PubMed
-
- Wortmann S.B., Duran M., Anikster Y., Barth P.G., Sperl W., Zschocke J., Morava E., Wevers R.A. Inborn errors of metabolism with 3-methylglutaconic aciduria as discriminative feature: proper classification and nomenclature. J. Inherit. Metab. Dis. 2013;36:923–928. - PubMed
-
- Wortmann S.B., Espeel M., Almeida L., Reimer A., Bosboom D., Roels F., de Brouwer A.P., Wevers R.A. Inborn errors of metabolism in the biosynthesis and remodelling of phospholipids. J. Inherit. Metab. Dis. 2014 Published online September 2, 2014. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
