

Novel variation and de novo mutation rates in population-wide de novo assembled Danish trios
- PMID: 25597990
- PMCID: PMC4309431
- DOI: 10.1038/ncomms6969
Novel variation and de novo mutation rates in population-wide de novo assembled Danish trios
Abstract
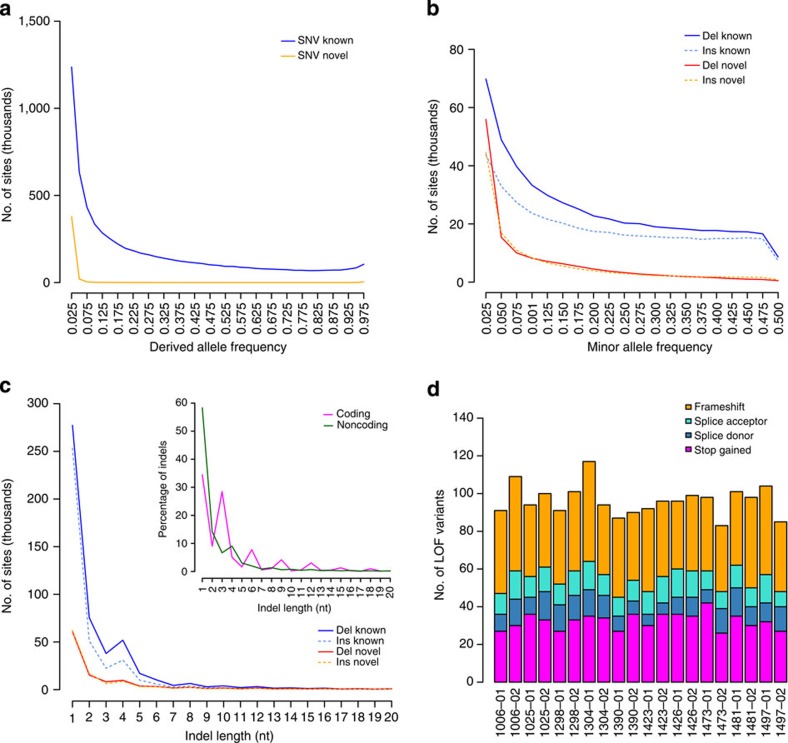
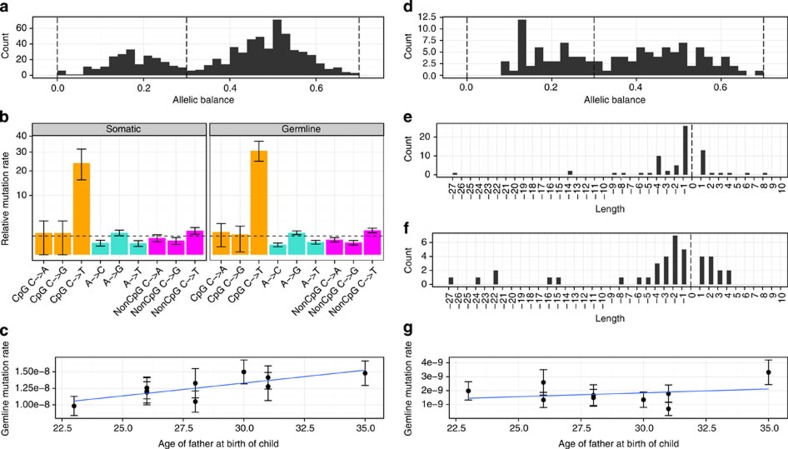
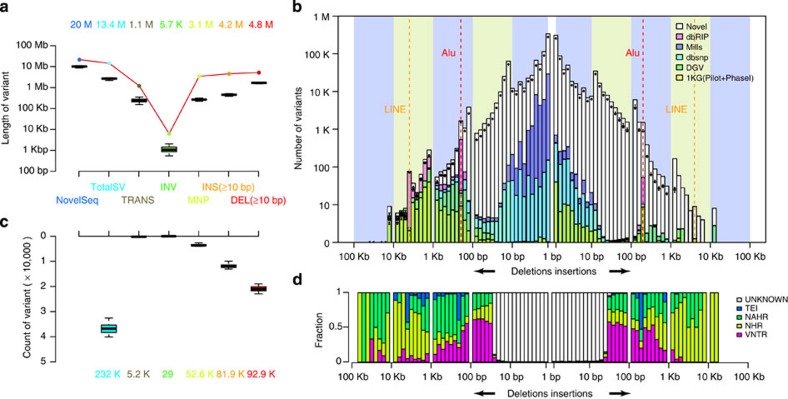
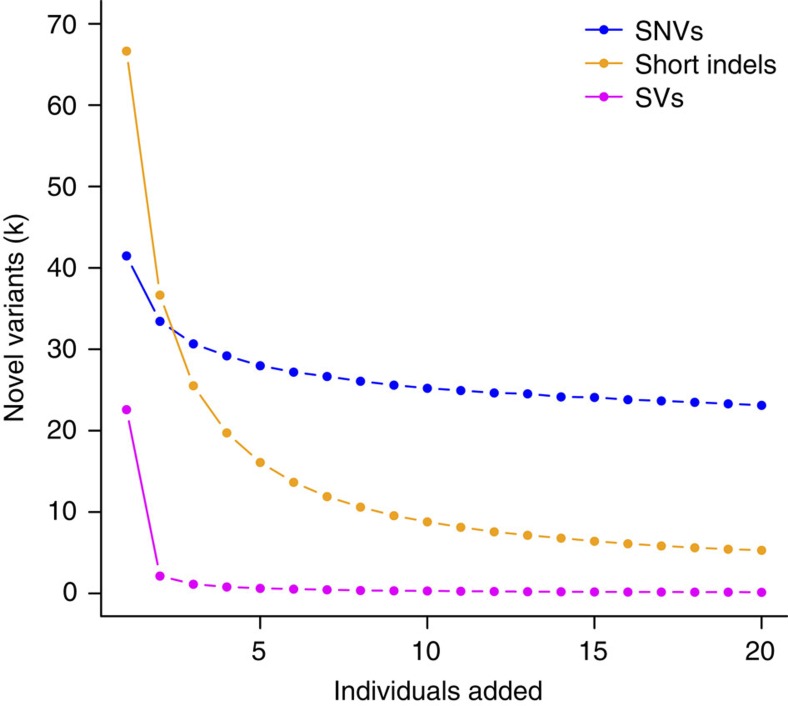
Building a population-specific catalogue of single nucleotide variants (SNVs), indels and structural variants (SVs) with frequencies, termed a national pan-genome, is critical for further advancing clinical and public health genetics in large cohorts. Here we report a Danish pan-genome obtained from sequencing 10 trios to high depth (50 × ). We report 536k novel SNVs and 283k novel short indels from mapping approaches and develop a population-wide de novo assembly approach to identify 132k novel indels larger than 10 nucleotides with low false discovery rates. We identify a higher proportion of indels and SVs than previous efforts showing the merits of high coverage and de novo assembly approaches. In addition, we use trio information to identify de novo mutations and use a probabilistic method to provide direct estimates of 1.27e-8 and 1.5e-9 per nucleotide per generation for SNVs and indels, respectively.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
