

A lipidomic screen of hyperglycemia-treated HRECs links 12/15-Lipoxygenase to microvascular dysfunction during diabetic retinopathy via NADPH oxidase
- PMID: 25598081
- PMCID: PMC4340307
- DOI: 10.1194/jlr.M056069
A lipidomic screen of hyperglycemia-treated HRECs links 12/15-Lipoxygenase to microvascular dysfunction during diabetic retinopathy via NADPH oxidase
Abstract
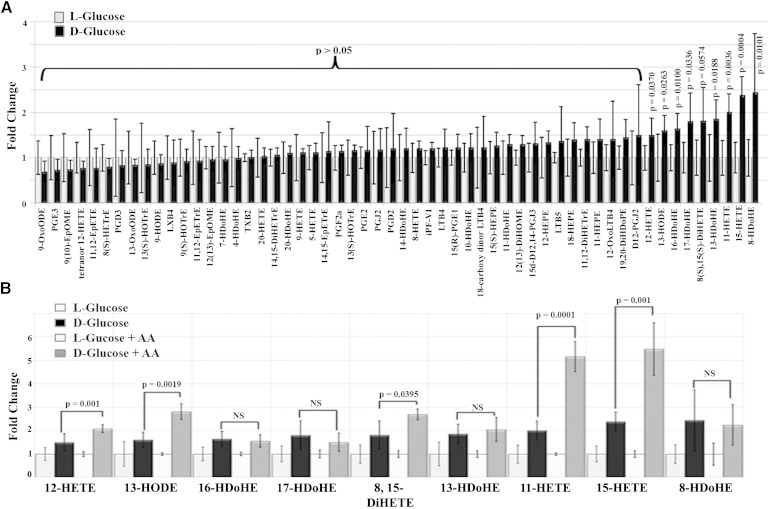
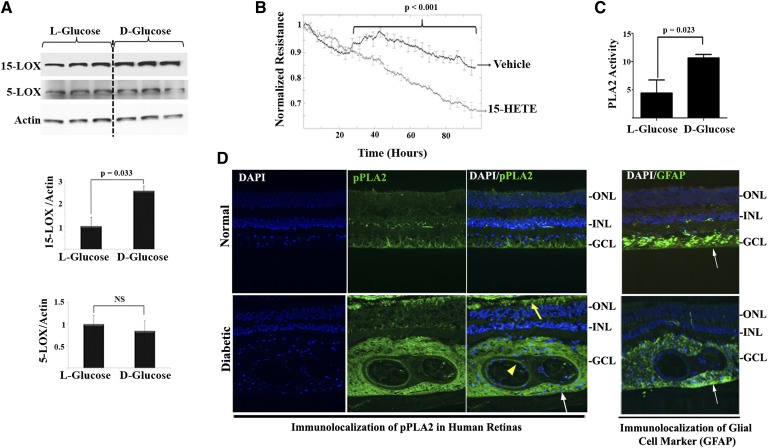
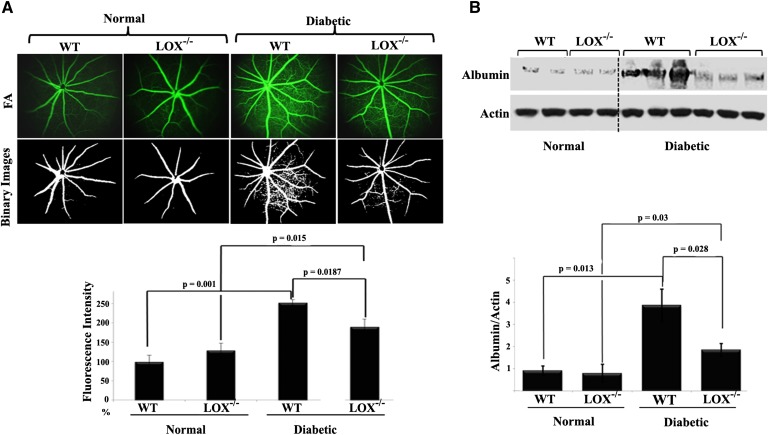
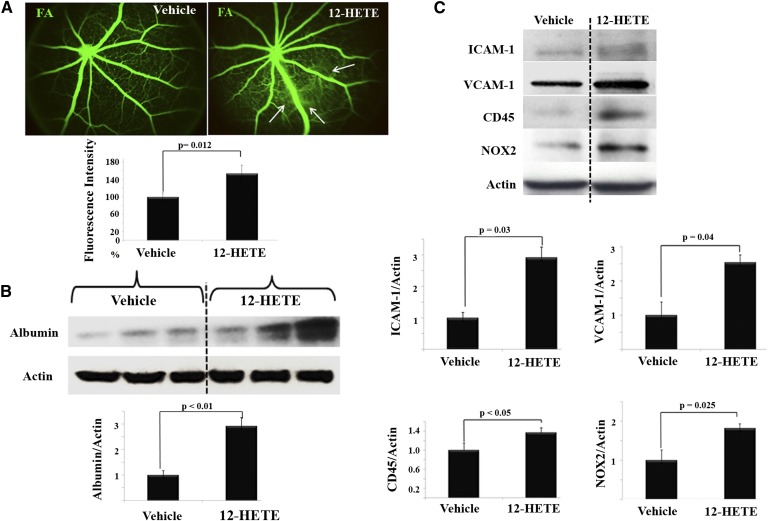
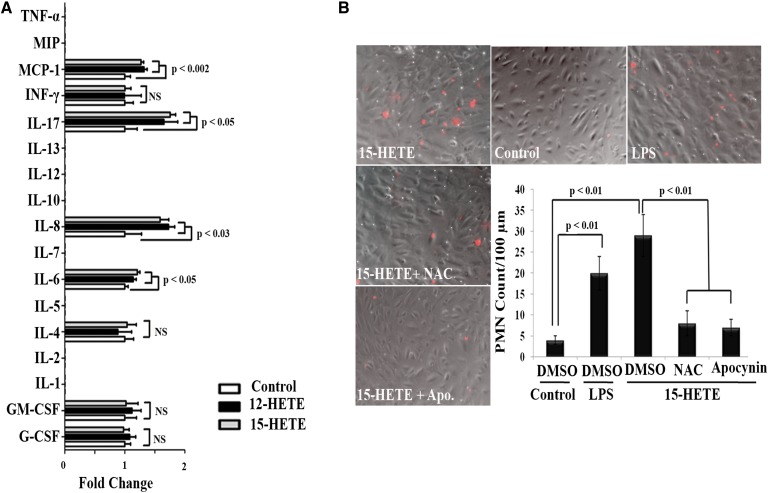
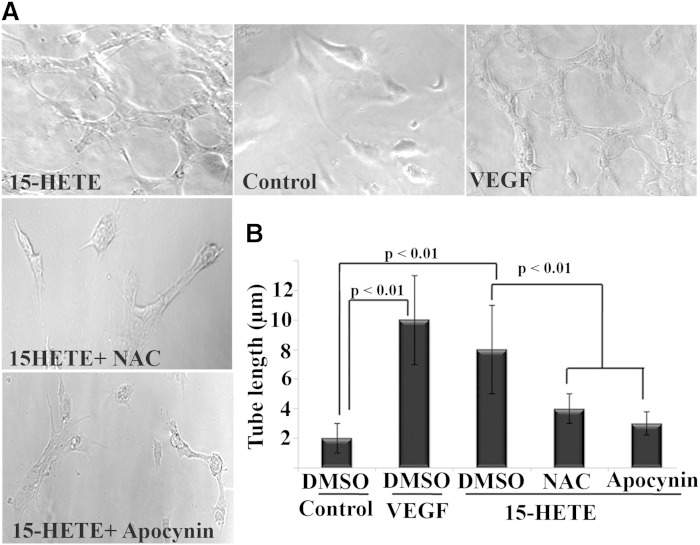
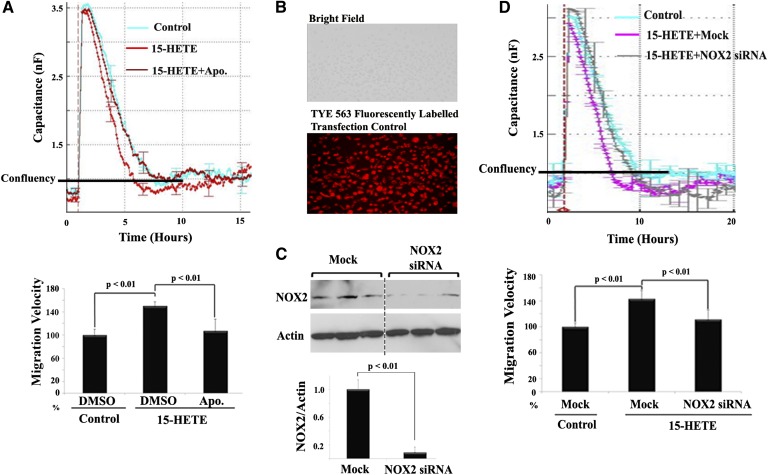
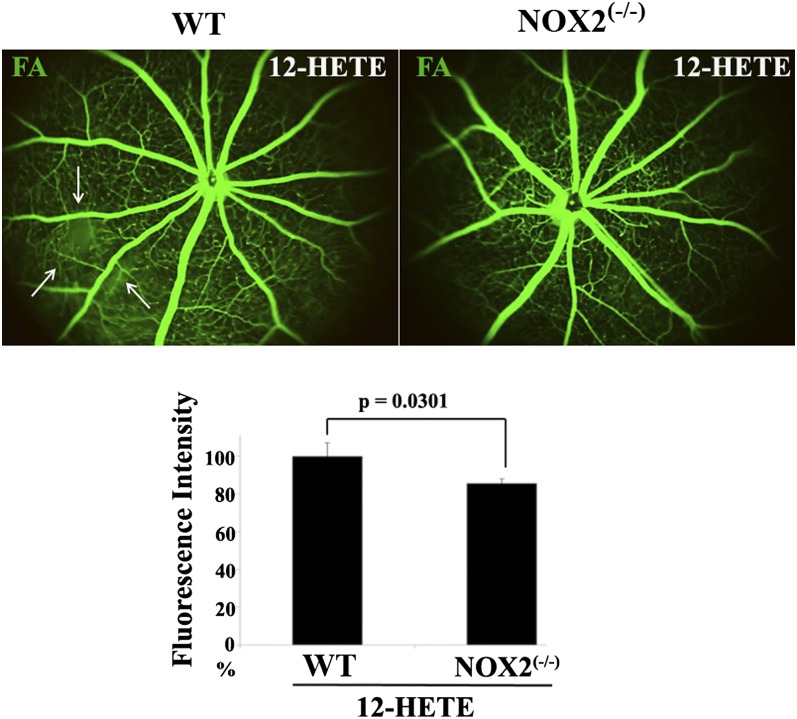
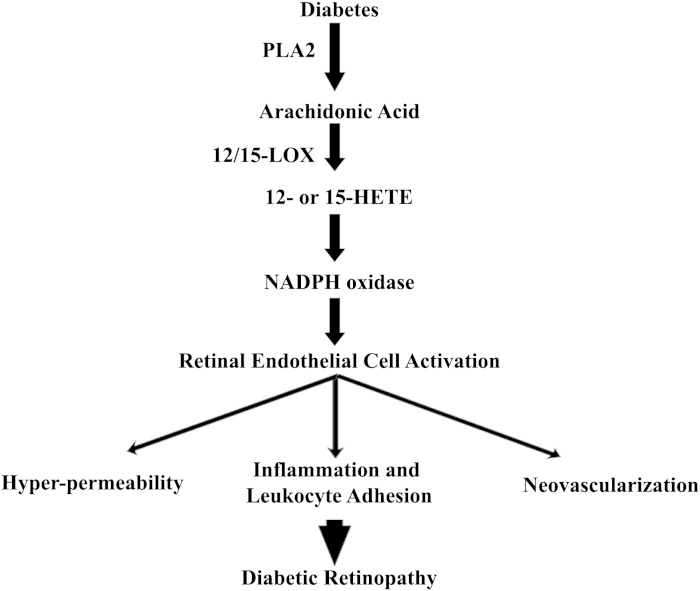
Retinal hyperpermeability and subsequent macular edema is a cardinal feature of early diabetic retinopathy (DR). Here, we investigated the role of bioactive lipid metabolites, in particular 12/15-lipoxygenase (LOX)-derived metabolites, in this process. LC/MS lipidomic screen of human retinal endothelial cells (HRECs) demonstrated that 15-HETE was the only significantly increased metabolite (2.4 ± 0.4-fold, P = 0.0004) by high glucose (30 mM) treatment. In the presence of arachidonic acid, additional eicosanoids generated by 12/15-LOX, including 12- and 11-HETEs, were significantly increased. Fluorescein angiography and retinal albumin leakage showed a significant decrease in retinal hyperpermeability in streptozotocin-induced diabetic mice lacking 12/15-LOX compared with diabetic WT mice. Our previous studies demonstrated the potential role of NADPH oxidase in mediating the permeability effect of 12- and 15-HETEs, therefore we tested the impact of intraocular injection of 12-HETE in mice lacking the catalytic subunit of NADPH oxidase (NOX2). The permeability effect of 12-HETE was significantly reduced in NOX2(-/-) mice compared with the WT mice. In vitro experiments also showed that 15-HETE induced HREC migration and tube formation in a NOX-dependent manner. Taken together our data suggest that 12/15-LOX is implicated in DR via a NOX-dependent mechanism.
Keywords: 12- and 15-HETEs; bioactive lipids; diabetic retinopathy; lipoxygenase; reduced nicotinamide adenine dinucleotide phosphate oxidase; retinal inflammation; retinal vascular leakage.
Copyright © 2015 by the American Society for Biochemistry and Molecular Biology, Inc.
Figures
Similar articles
-
Pigment epithelium-derived factor inhibits retinal microvascular dysfunction induced by 12/15-lipoxygenase-derived eicosanoids.Biochim Biophys Acta. 2015 Mar;1851(3):290-8. doi: 10.1016/j.bbalip.2014.12.017. Epub 2015 Jan 3. Biochim Biophys Acta. 2015. PMID: 25562624 Free PMC article.
-
12/15-Lipoxygenase-derived lipid metabolites induce retinal endothelial cell barrier dysfunction: contribution of NADPH oxidase.PLoS One. 2013;8(2):e57254. doi: 10.1371/journal.pone.0057254. Epub 2013 Feb 20. PLoS One. 2013. PMID: 23437353 Free PMC article.
-
Role of endoplasmic reticulum stress in 12/15-lipoxygenase-induced retinal microvascular dysfunction in a mouse model of diabetic retinopathy.Diabetologia. 2018 May;61(5):1220-1232. doi: 10.1007/s00125-018-4560-z. Epub 2018 Feb 21. Diabetologia. 2018. PMID: 29468369 Free PMC article.
-
Key Role of 12-Lipoxygenase and Its Metabolite 12-Hydroxyeicosatetraenoic Acid (12-HETE) in Diabetic Retinopathy.Curr Eye Res. 2022 Mar;47(3):329-335. doi: 10.1080/02713683.2021.1995003. Epub 2022 Feb 7. Curr Eye Res. 2022. PMID: 35129022 Review.
-
Diabetic retinopathy: Focus on NADPH oxidase and its potential as therapeutic target.Eur J Pharmacol. 2019 Jun 15;853:381-387. doi: 10.1016/j.ejphar.2019.04.038. Epub 2019 Apr 19. Eur J Pharmacol. 2019. PMID: 31009636 Review.
Cited by
-
Hyperhomocysteinemia dysregulates plasma levels of polyunsaturated fatty acids-derived eicosanoids.Life Res (Auckl). 2022 Apr;5(2):14. doi: 10.53388/2022-0106-103. Life Res (Auckl). 2022. PMID: 36341141 Free PMC article.
-
Effects of Arachidonic Acid Metabolites on Cardiovascular Health and Disease.Int J Mol Sci. 2021 Nov 6;22(21):12029. doi: 10.3390/ijms222112029. Int J Mol Sci. 2021. PMID: 34769460 Free PMC article. Review.
-
Spermine oxidase: A promising therapeutic target for neurodegeneration in diabetic retinopathy.Pharmacol Res. 2019 Sep;147:104299. doi: 10.1016/j.phrs.2019.104299. Epub 2019 Jun 15. Pharmacol Res. 2019. PMID: 31207342 Free PMC article. Review.
-
A novel interaction between soluble epoxide hydrolase and the AT1 receptor in retinal microvascular damage.Prostaglandins Other Lipid Mediat. 2020 Jun;148:106449. doi: 10.1016/j.prostaglandins.2020.106449. Epub 2020 Apr 30. Prostaglandins Other Lipid Mediat. 2020. PMID: 32360774 Free PMC article.
-
Inhibition of 12/15-Lipoxygenase Reduces Renal Inflammation and Injury in Streptozotocin-Induced Diabetic Mice.J Diabetes Metab. 2015 Jun;6(6):555. doi: 10.4172/2155-6156.1000555. Epub 2015 May 30. J Diabetes Metab. 2015. PMID: 26823989 Free PMC article.
References
-
- Fong D. S., Aiello L. P., Ferris F. L., III, Klein R. 2004. Diabetic retinopathy. Diabetes Care. 27: 2540–2553. - PubMed
-
- Frank R. N. 2004. Diabetic retinopathy. N. Engl. J. Med. 350: 48–58. - PubMed
-
- Jeganathan V. S. 2011. Anti-angiogenesis drugs in diabetic retinopathy. Curr. Pharm. Biotechnol. 12: 369–372. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
