

The landscape of long noncoding RNAs in the human transcriptome
- PMID: 25599403
- PMCID: PMC4417758
- DOI: 10.1038/ng.3192
The landscape of long noncoding RNAs in the human transcriptome
Abstract
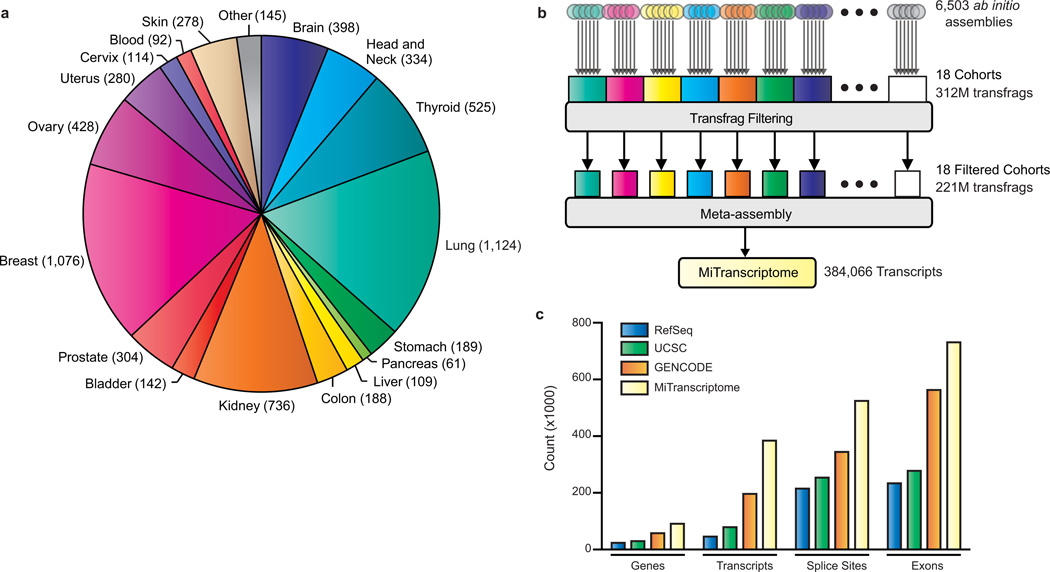
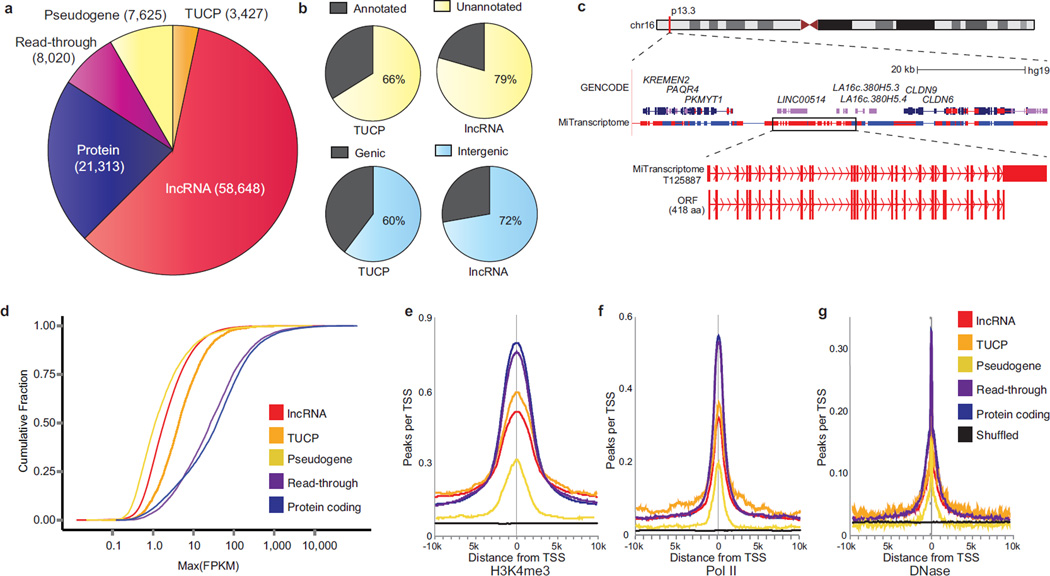
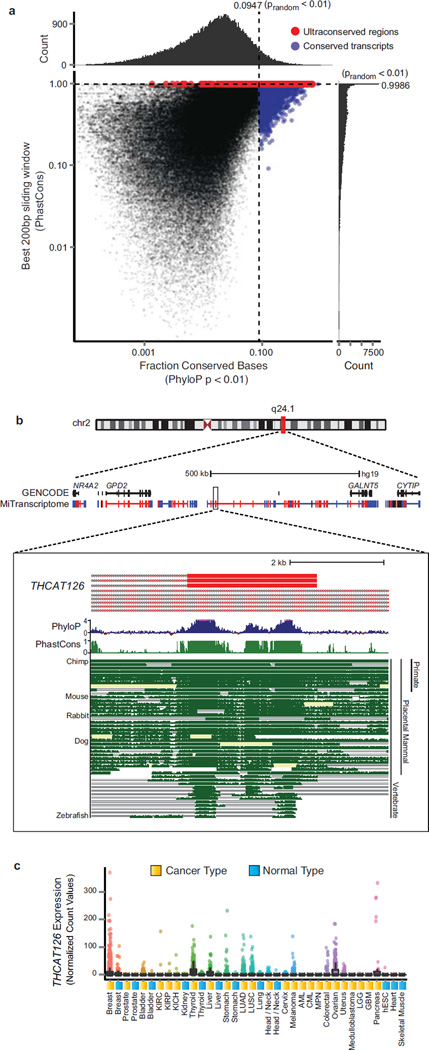
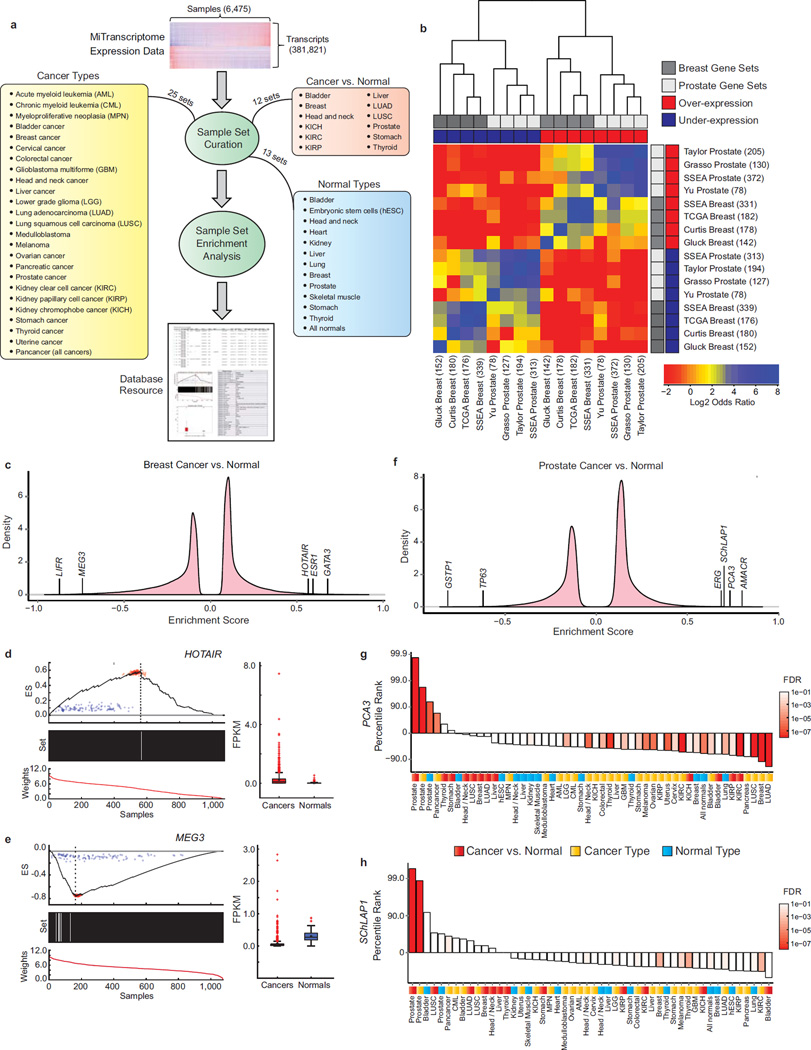
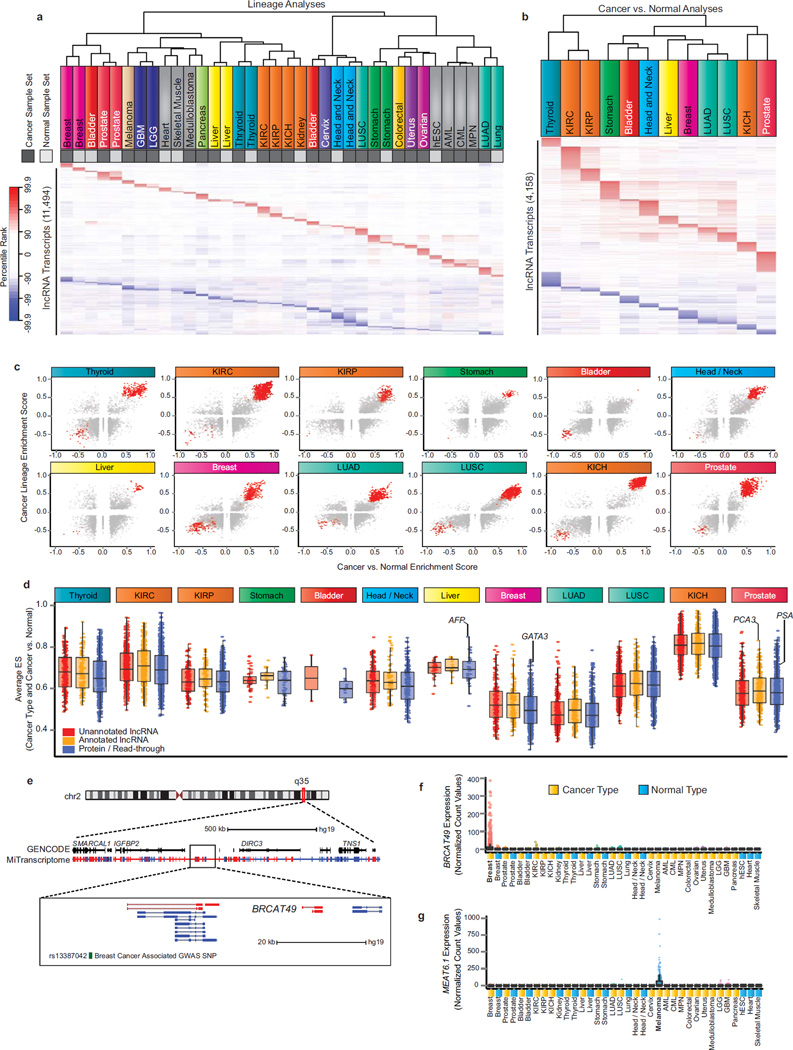
Long noncoding RNAs (lncRNAs) are emerging as important regulators of tissue physiology and disease processes including cancer. To delineate genome-wide lncRNA expression, we curated 7,256 RNA sequencing (RNA-seq) libraries from tumors, normal tissues and cell lines comprising over 43 Tb of sequence from 25 independent studies. We applied ab initio assembly methodology to this data set, yielding a consensus human transcriptome of 91,013 expressed genes. Over 68% (58,648) of genes were classified as lncRNAs, of which 79% were previously unannotated. About 1% (597) of the lncRNAs harbored ultraconserved elements, and 7% (3,900) overlapped disease-associated SNPs. To prioritize lineage-specific, disease-associated lncRNA expression, we employed non-parametric differential expression testing and nominated 7,942 lineage- or cancer-associated lncRNA genes. The lncRNA landscape characterized here may shed light on normal biology and cancer pathogenesis and may be valuable for future biomarker development.
Figures
References
-
- Ferlay J, et al. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. International Journal of Cancer. 2014 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
