

DIA-Umpire: comprehensive computational framework for data-independent acquisition proteomics
- PMID: 25599550
- PMCID: PMC4399776
- DOI: 10.1038/nmeth.3255
DIA-Umpire: comprehensive computational framework for data-independent acquisition proteomics
Abstract
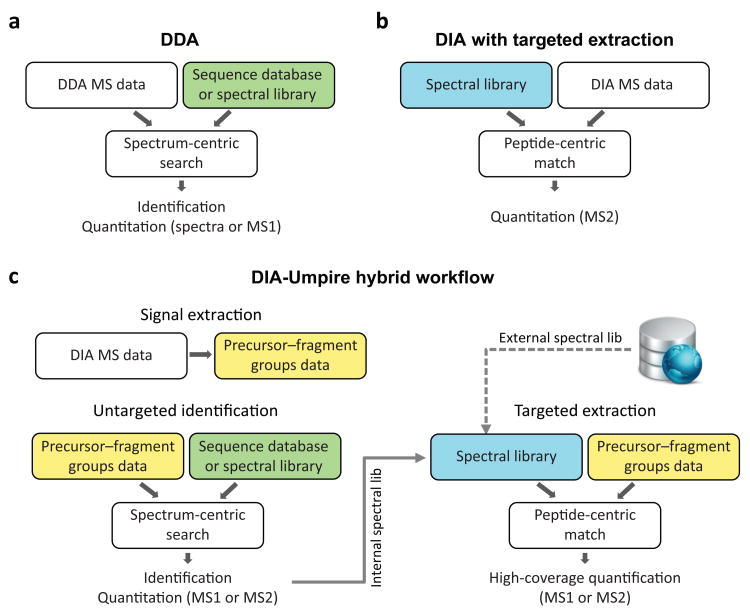
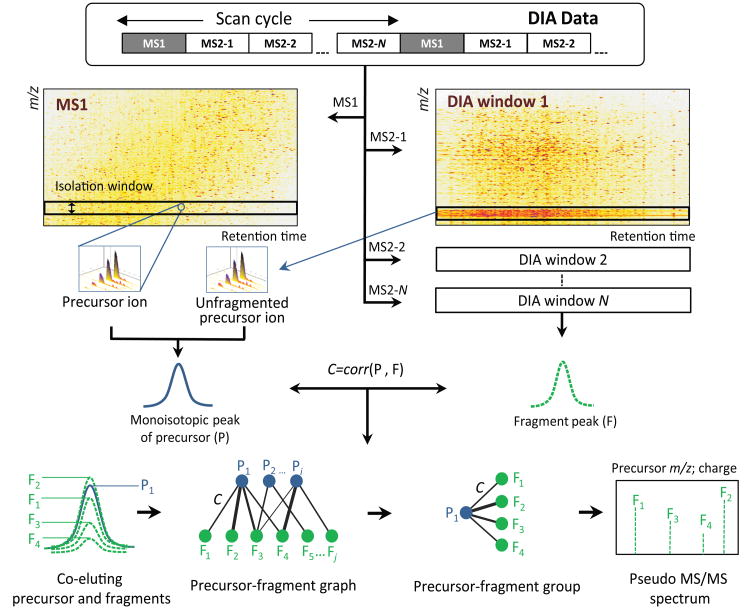
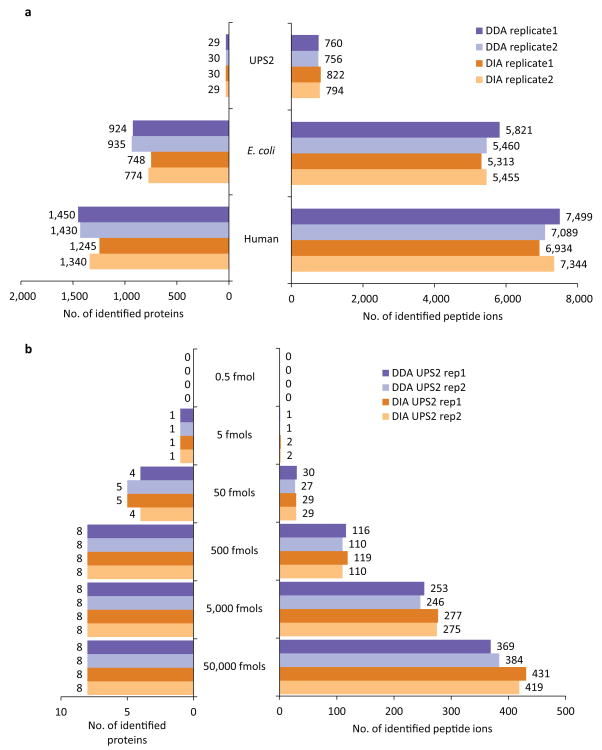
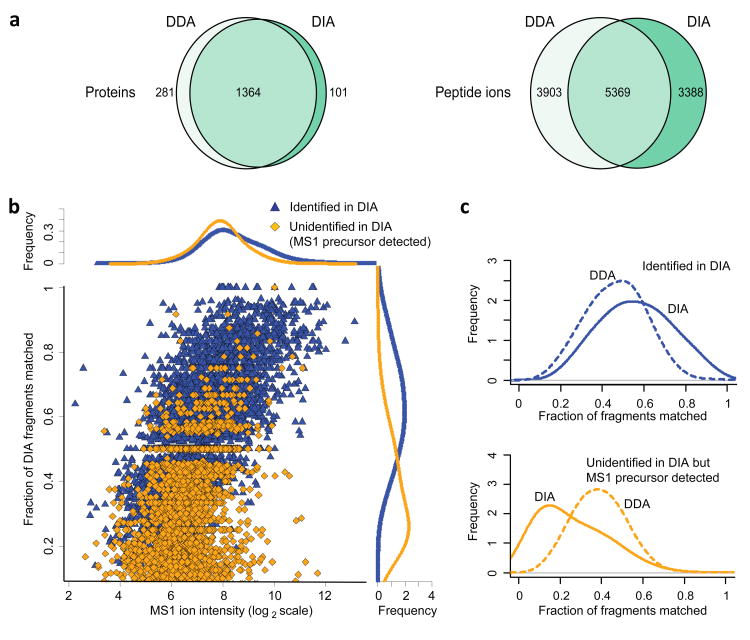
As a result of recent improvements in mass spectrometry (MS), there is increased interest in data-independent acquisition (DIA) strategies in which all peptides are systematically fragmented using wide mass-isolation windows ('multiplex fragmentation'). DIA-Umpire (http://diaumpire.sourceforge.net/), a comprehensive computational workflow and open-source software for DIA data, detects precursor and fragment chromatographic features and assembles them into pseudo-tandem MS spectra. These spectra can be identified with conventional database-searching and protein-inference tools, allowing sensitive, untargeted analysis of DIA data without the need for a spectral library. Quantification is done with both precursor- and fragment-ion intensities. Furthermore, DIA-Umpire enables targeted extraction of quantitative information based on peptides initially identified in only a subset of the samples, resulting in more consistent quantification across multiple samples. We demonstrated the performance of the method with control samples of varying complexity and publicly available glycoproteomics and affinity purification-MS data.
Figures
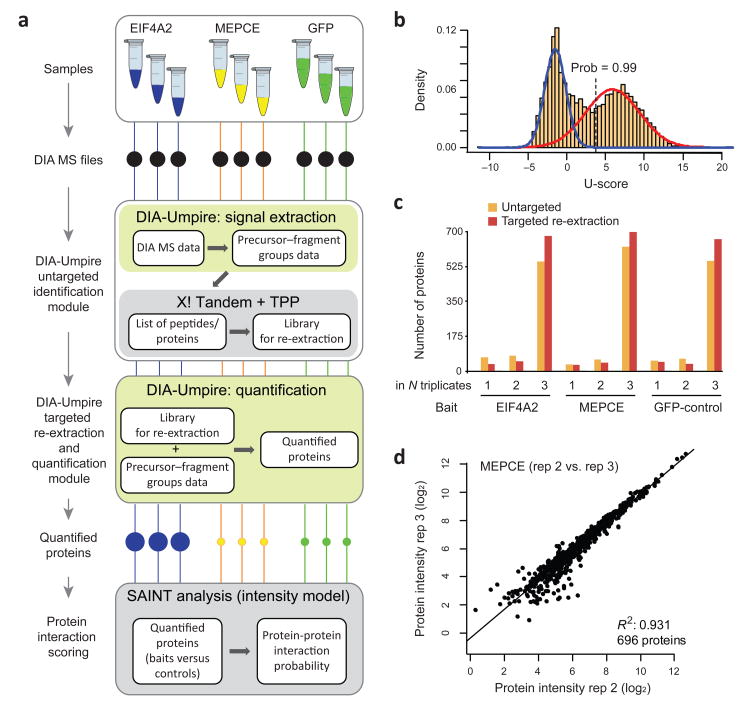
References
-
- Bantscheff M, Lemeer S, Savitski MM, Kuster B. Quantitative mass spectrometry in proteomics: critical review update from 2007 to the present. Anal Bioanal Chem. 2012;404:939–965. - PubMed
-
- Michalski A, Cox J, Mann M. More than 100,000 Detectable Peptide Species Elute in Single Shotgun Proteomics Runs but the Majority is Inaccessible to Data-Dependent LC-MS/MS. J Proteome Res. 2011;10:1785–1793. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
