

Churchill: an ultra-fast, deterministic, highly scalable and balanced parallelization strategy for the discovery of human genetic variation in clinical and population-scale genomics
- PMID: 25600152
- PMCID: PMC4333267
- DOI: 10.1186/s13059-014-0577-x
Churchill: an ultra-fast, deterministic, highly scalable and balanced parallelization strategy for the discovery of human genetic variation in clinical and population-scale genomics
Abstract
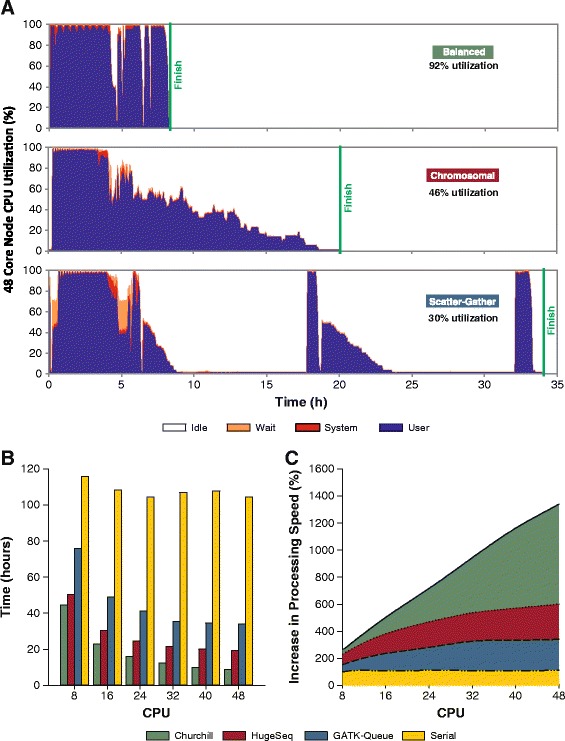
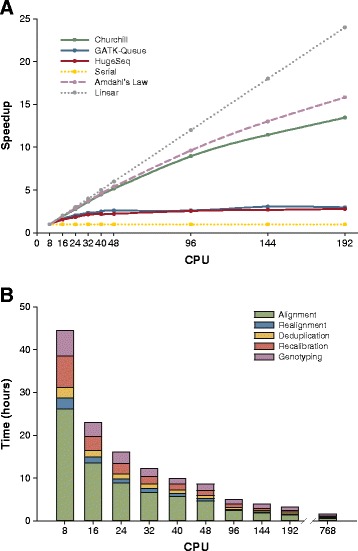
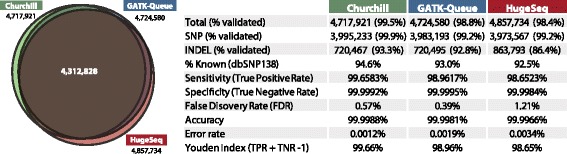
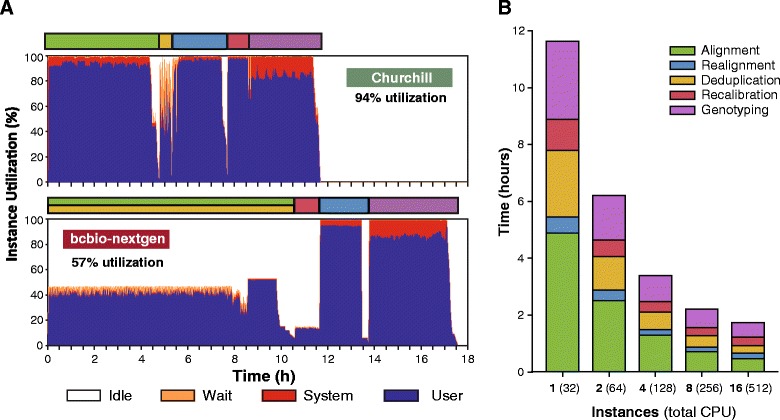
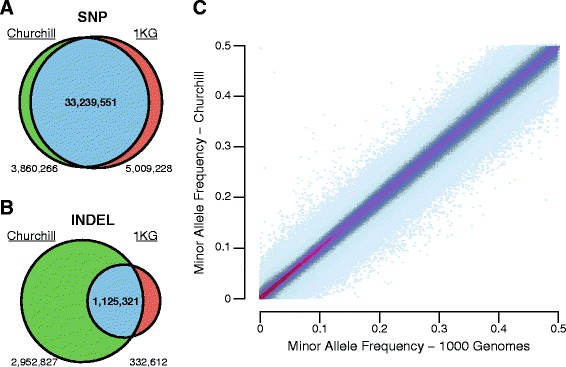
While advances in genome sequencing technology make population-scale genomics a possibility, current approaches for analysis of these data rely upon parallelization strategies that have limited scalability, complex implementation and lack reproducibility. Churchill, a balanced regional parallelization strategy, overcomes these challenges, fully automating the multiple steps required to go from raw sequencing reads to variant discovery. Through implementation of novel deterministic parallelization techniques, Churchill allows computationally efficient analysis of a high-depth whole genome sample in less than two hours. The method is highly scalable, enabling full analysis of the 1000 Genomes raw sequence dataset in a week using cloud resources. http://churchill.nchri.org/.
Figures
References
-
- The Boston Children’s Hospital CLARITY Challenge Consortium An international effort towards developing standards for best practices in analysis, interpretation and reporting of clinical genome sequencing results in the CLARITY Challenge. Genome Biol. 2014;15:R53. doi: 10.1186/gb-2014-15-3-r53. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
