

Tunable-combinatorial mechanisms of acquired resistance limit the efficacy of BRAF/MEK cotargeting but result in melanoma drug addiction
- PMID: 25600339
- PMCID: PMC4326539
- DOI: 10.1016/j.ccell.2014.11.018
Tunable-combinatorial mechanisms of acquired resistance limit the efficacy of BRAF/MEK cotargeting but result in melanoma drug addiction
Abstract
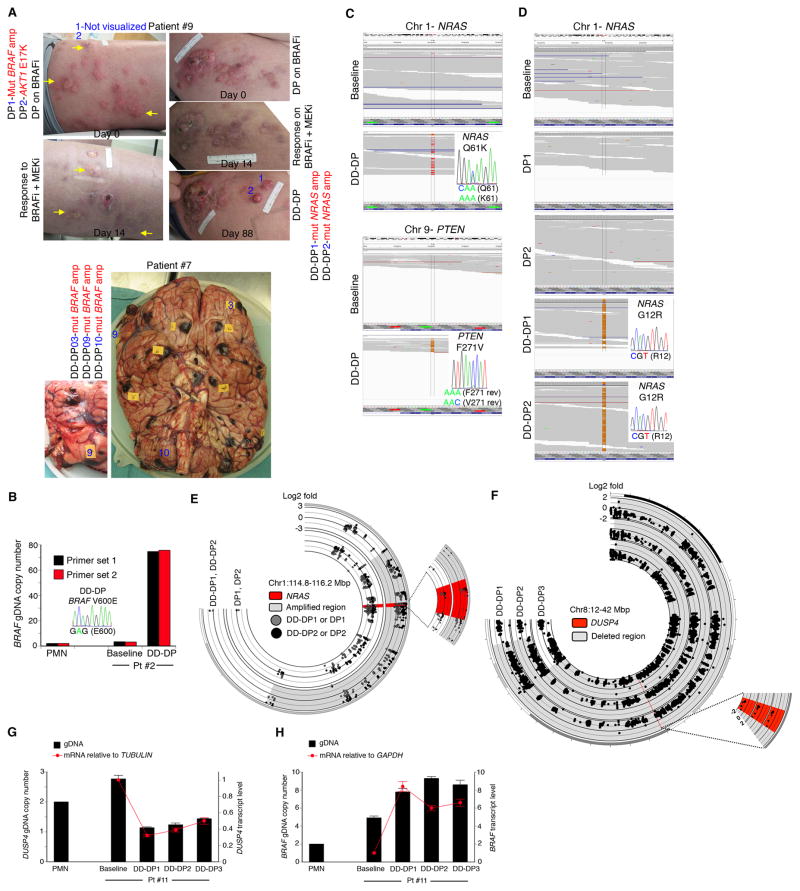
Combined BRAF- and MEK-targeted therapy improves upon BRAF inhibitor (BRAFi) therapy but is still beset by acquired resistance. We show that melanomas acquire resistance to combined BRAF and MEK inhibition by augmenting or combining mechanisms of single-agent BRAFi resistance. These double-drug resistance-associated genetic configurations significantly altered molecular interactions underlying MAPK pathway reactivation. (V600E)BRAF, expressed at supraphysiological levels because of (V600E)BRAF ultra-amplification, dimerized with and activated CRAF. In addition, MEK mutants enhanced interaction with overexpressed (V600E)BRAF via a regulatory interface at R662 of (V600E)BRAF. Importantly, melanoma cell lines selected for resistance to BRAFi+MEKi, but not those to BRAFi alone, displayed robust drug addiction, providing a potentially exploitable therapeutic opportunity.
Copyright © 2015 Elsevier Inc. All rights reserved.
Conflict of interest statement
No author declared conflicts of interest.
Figures







Comment in
-
Targeted therapies: Drug addiction revealed in BRAF and MEK inhibitor-resistant melanoma cells.Nat Rev Clin Oncol. 2015 Apr;12(4):189. doi: 10.1038/nrclinonc.2015.28. Epub 2015 Feb 17. Nat Rev Clin Oncol. 2015. PMID: 25687909 No abstract available.
References
-
- Brennan DF, Dar AC, Hertz NT, Chao WC, Burlingame AL, Shokat KM, Barford D. A Raf-induced allosteric transition of KSR stimulates phosphorylation of MEK. Nature. 2011;472:366–369. - PubMed
-
- Catalanotti F, Reyes G, Jesenberger V, Galabova-Kovacs G, de Matos Simoes R, Carugo O, Baccarini M. A Mek1-Mek2 heterodimer determines the strength and duration of the Erk signal. Nat Struct Mol Biol. 2009;16:294–303. - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
