

Cancer systems biology of TCGA SKCM: efficient detection of genomic drivers in melanoma
- PMID: 25600636
- PMCID: PMC4298731
- DOI: 10.1038/srep07857
Cancer systems biology of TCGA SKCM: efficient detection of genomic drivers in melanoma
Abstract
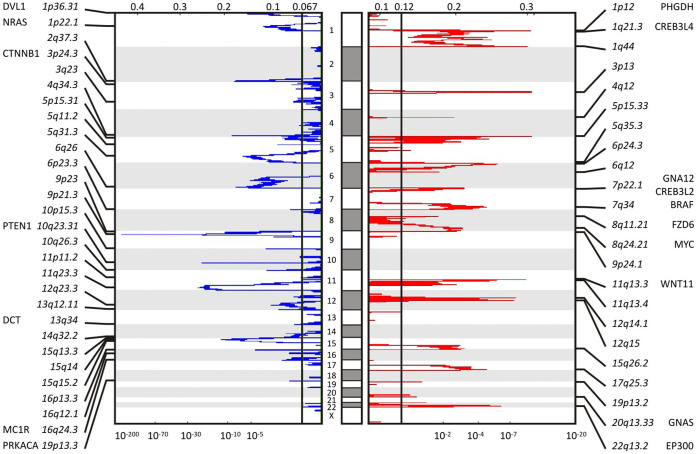
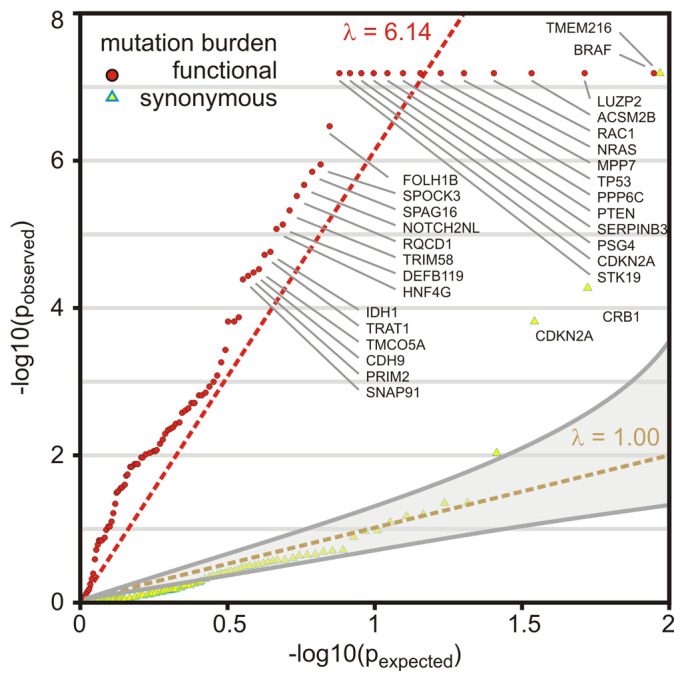
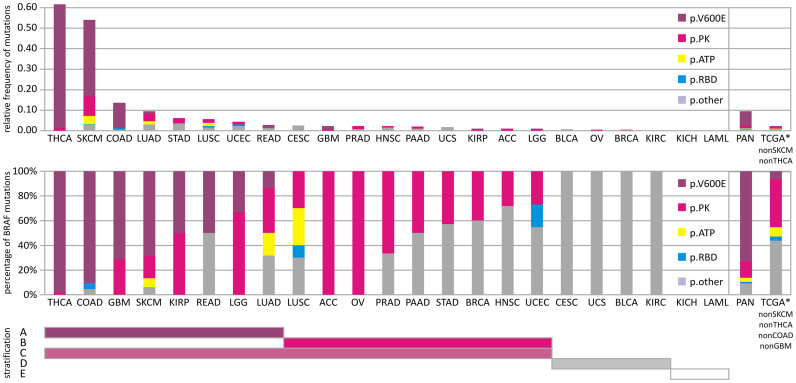
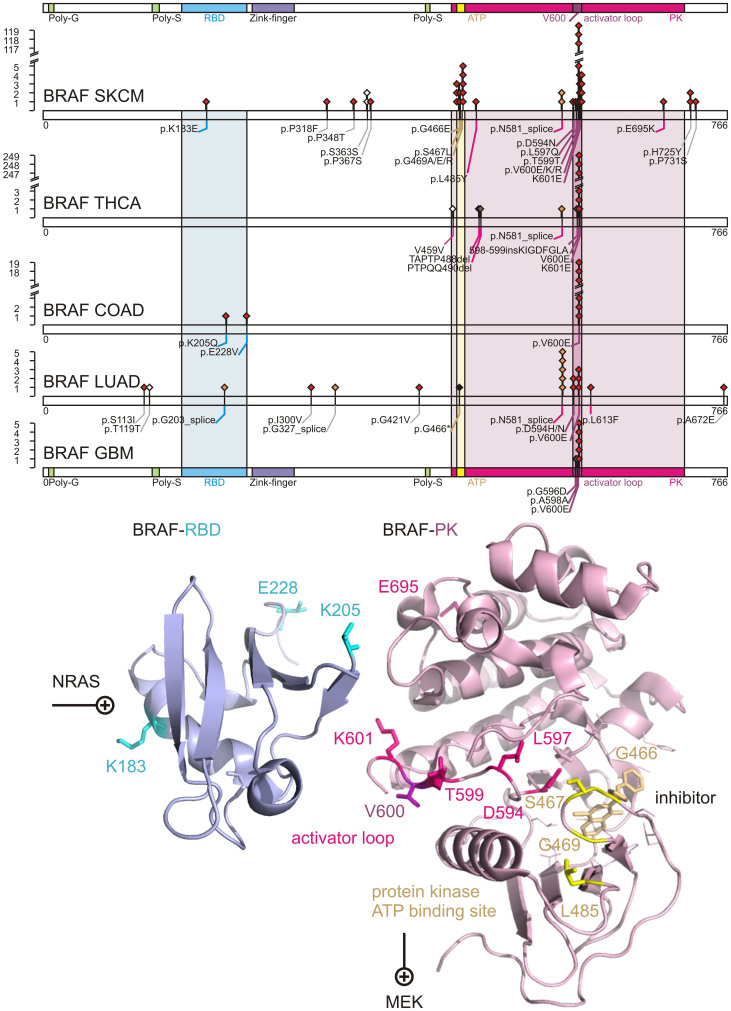
We characterized the mutational landscape of human skin cutaneous melanoma (SKCM) using data obtained from The Cancer Genome Atlas (TCGA) project. We analyzed next-generation sequencing data of somatic copy number alterations and somatic mutations in 303 metastatic melanomas. We were able to confirm preeminent drivers of melanoma as well as identify new melanoma genes. The TCGA SKCM study confirmed a dominance of somatic BRAF mutations in 50% of patients. The mutational burden of melanoma patients is an order of magnitude higher than of other TCGA cohorts. A multi-step filter enriched somatic mutations while accounting for recurrence, conservation, and basal rate. Thus, this filter can serve as a paradigm for analysis of genome-wide next-generation sequencing data of large cohorts with a high mutational burden. Analysis of TCGA melanoma data using such a multi-step filter discovered novel and statistically significant potential melanoma driver genes. In the context of the Pan-Cancer study we report a detailed analysis of the mutational landscape of BRAF and other drivers across cancer tissues. Integrated analysis of somatic mutations, somatic copy number alterations, low pass copy numbers, and gene expression of the melanogenesis pathway shows coordination of proliferative events by Gs-protein and cyclin signaling at a systems level.
Figures
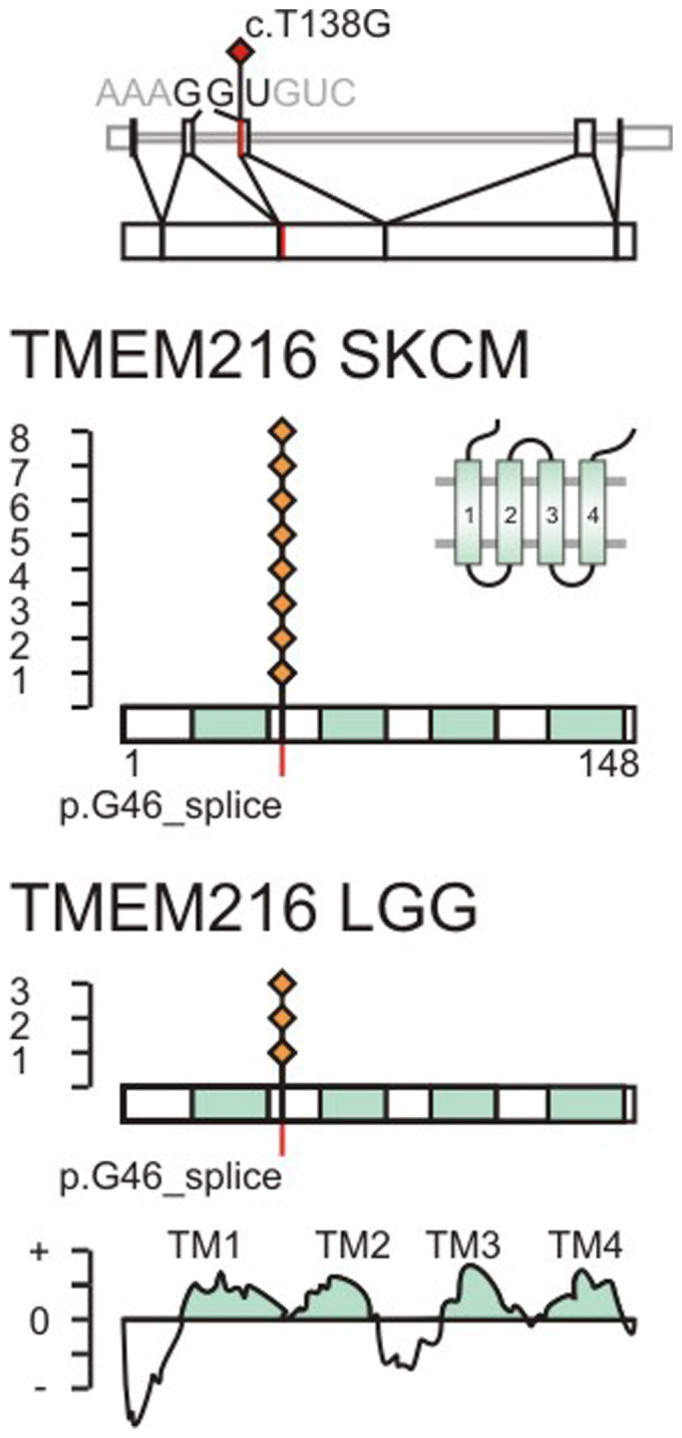
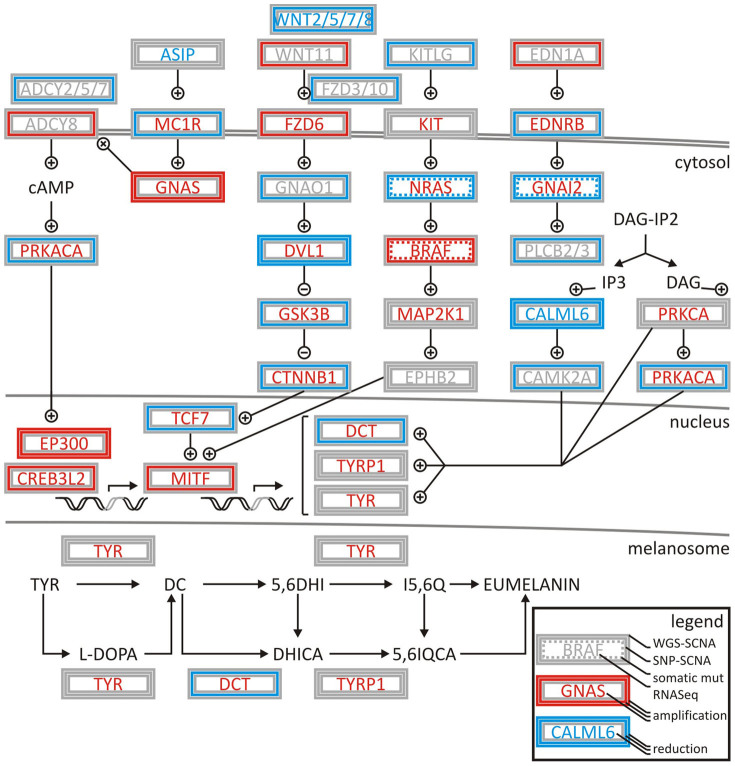
References
-
- Meyerson M., Gabriel S. & Getz G. Advances in understanding cancer genomes through second-generation sequencing. Nat Rev Genet 11, 685–696 (2010). - PubMed
-
- Davies H. et al. Mutations of the BRAF gene in human cancer. Nature 417, 949–954 (2002). - PubMed
-
- Pollock P. M. et al. High frequency of BRAF mutations in nevi. Nat Genet 33, 19–20 (2003). - PubMed
-
- Flaherty K. T. et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med 367, 107–114 (2012). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
