

Cooperation between Noncanonical Ras Network Mutations
- PMID: 25600866
- PMCID: PMC4503519
- DOI: 10.1016/j.celrep.2014.12.035
Cooperation between Noncanonical Ras Network Mutations
Erratum in
-
Cooperation between Noncanonical Ras Network Mutations.Cell Rep. 2015 Feb 10;10(5):840. doi: 10.1016/j.celrep.2015.01.048. Epub 2015 Feb 11. Cell Rep. 2015. PMID: 30849858 No abstract available.
Abstract
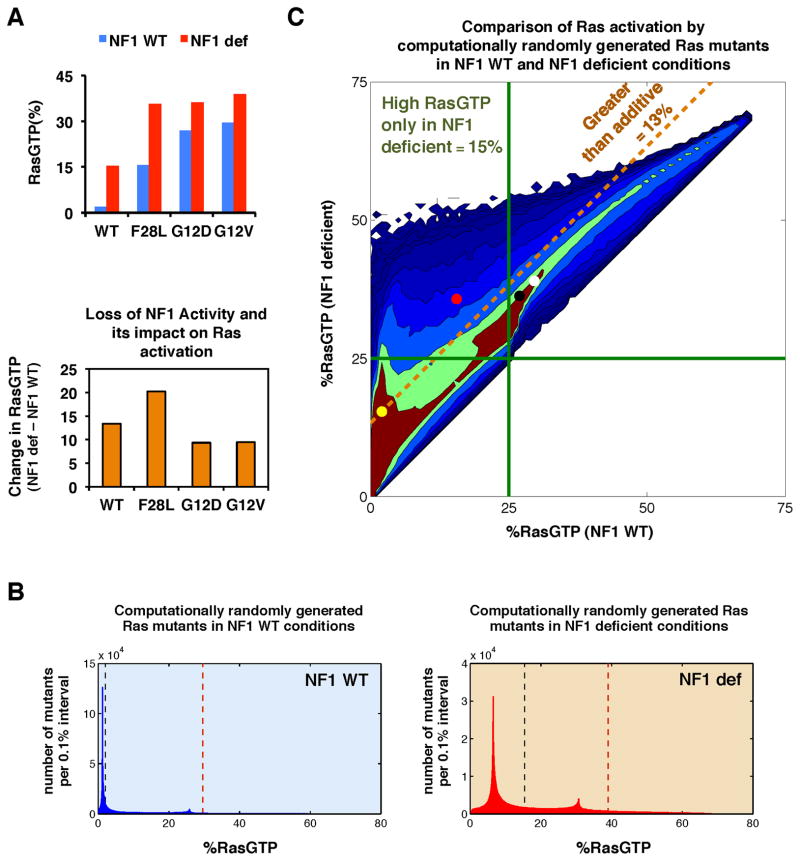
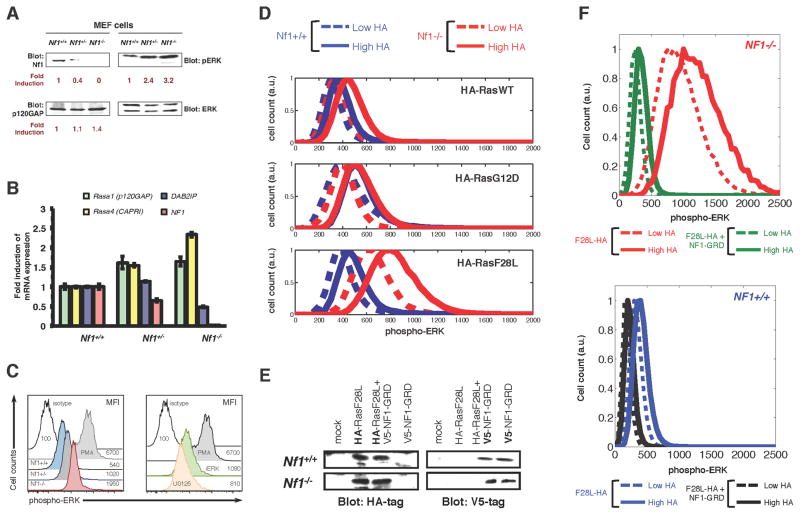
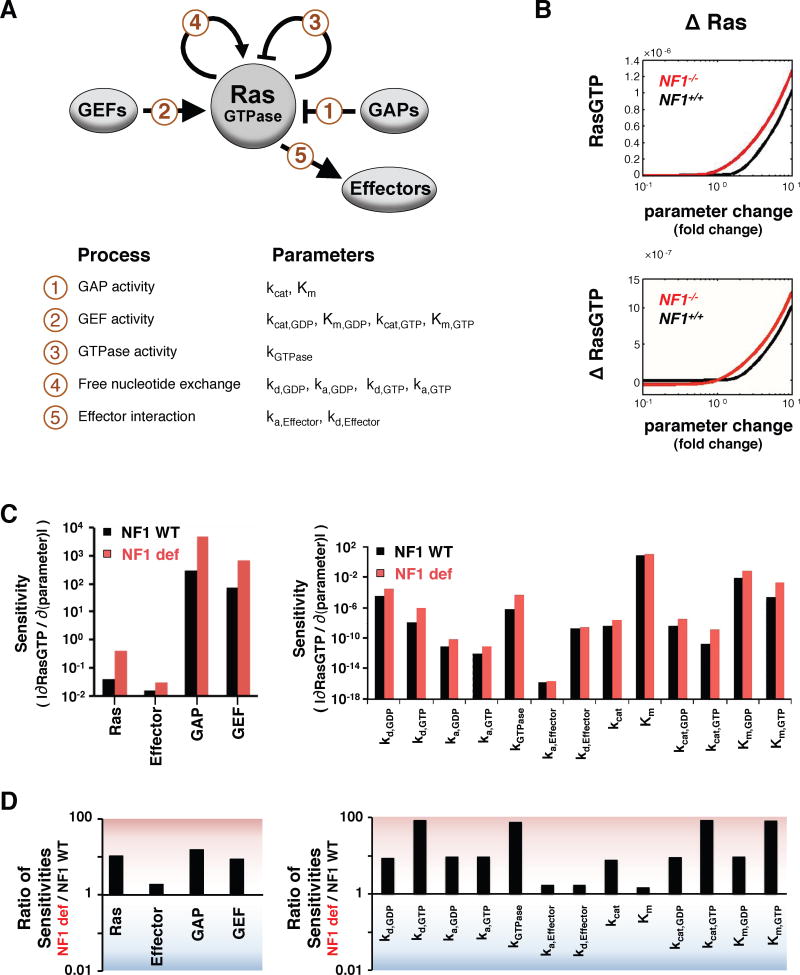
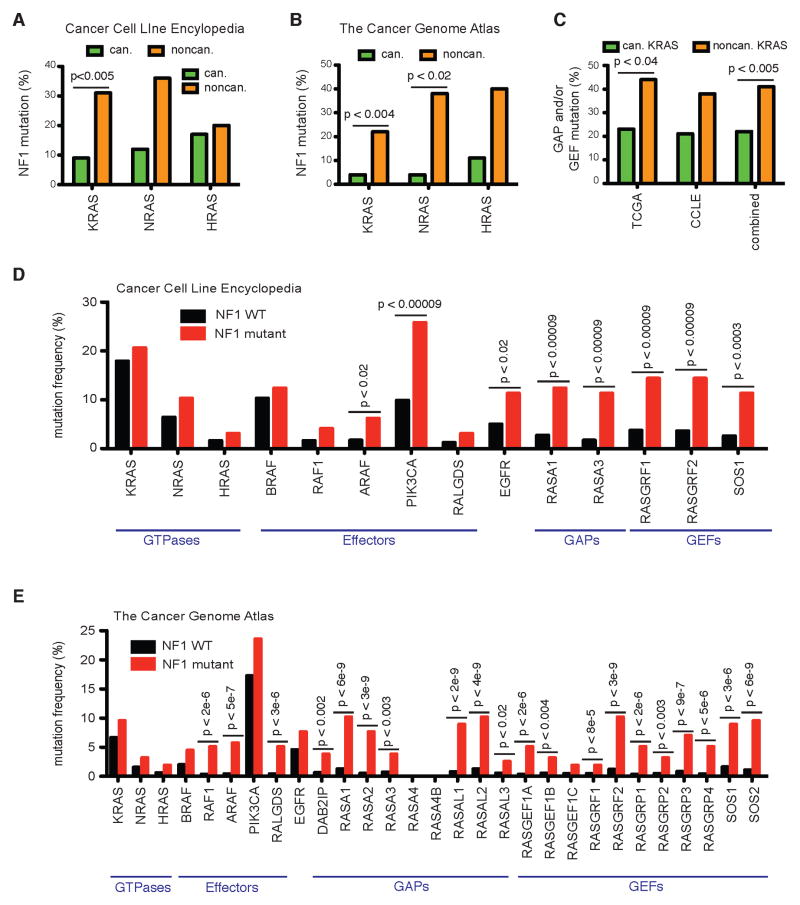
Cancer develops after the acquisition of a collection of mutations that together create the cancer phenotype. How collections of mutations work together within a cell and whether there is selection for certain combinations of mutations are not well understood. We investigated this problem with a mathematical model of the Ras signaling network, including a computational random mutagenesis. Modeling and subsequent experiments revealed that mutations of the tumor suppressor gene NF1 can amplify the effects of other Ras pathway mutations, including weakly activating, noncanonical Ras mutants. Furthermore, analyzing recently available, large, cancer genomic data sets uncovered increased co-occurrence of NF1 mutations with mutations in other Ras network genes. Overall, these data suggest that combinations of Ras pathway mutations could serve the role of cancer "driver." More generally, this work suggests that mutations that result in network instability may promote cancer in a manner analogous to genomic instability.
Copyright © 2015 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Beckman RA, Loeb LA. Genetic instability in cancer: theory and experiment. Semin Cancer Biol. 2005;15:423–435. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
