

Peaks cloaked in the mist: the landscape of mammalian replication origins
- PMID: 25601401
- PMCID: PMC4298691
- DOI: 10.1083/jcb.201407004
Peaks cloaked in the mist: the landscape of mammalian replication origins
Abstract
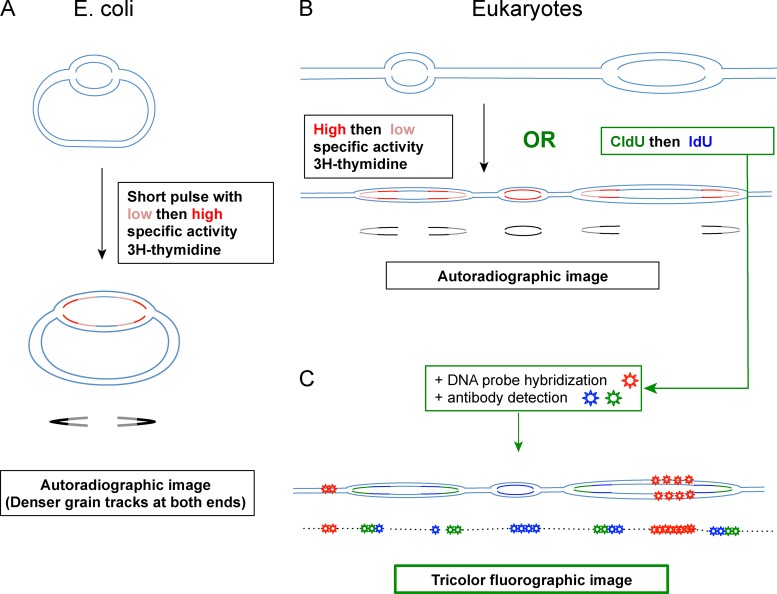
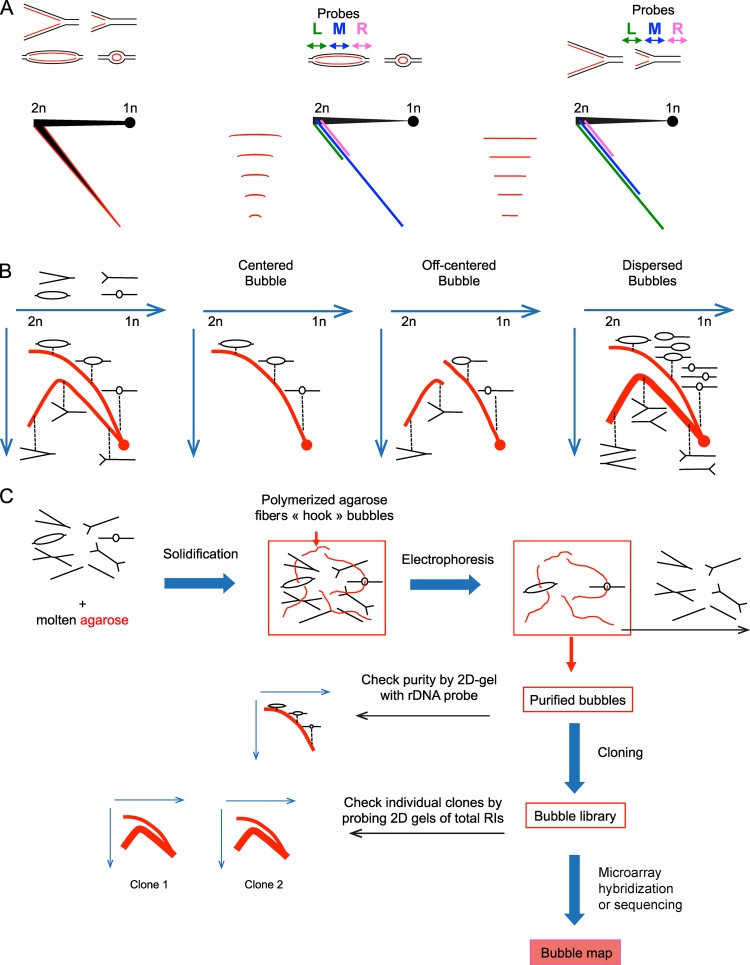
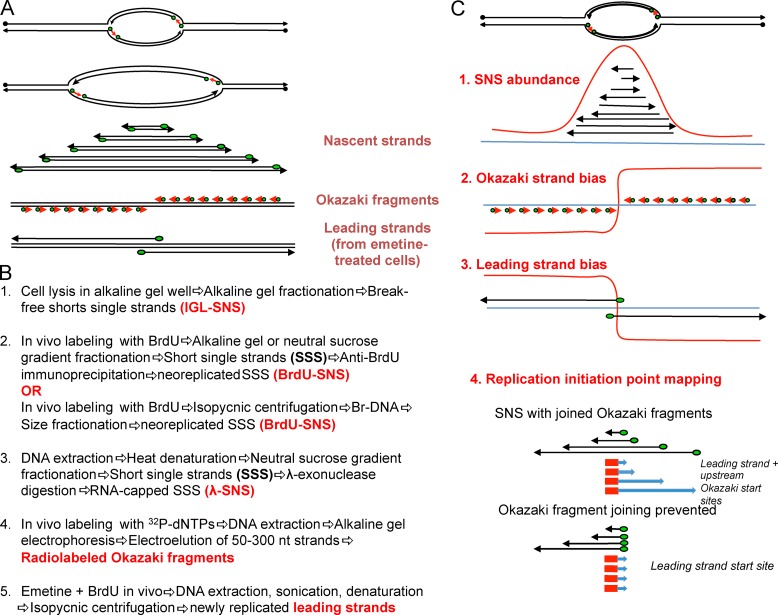
Replication of mammalian genomes starts at sites termed replication origins, which historically have been difficult to locate as a result of large genome sizes, limited power of genetic identification schemes, and rareness and fragility of initiation intermediates. However, origins are now mapped by the thousands using microarrays and sequencing techniques. Independent studies show modest concordance, suggesting that mammalian origins can form at any DNA sequence but are suppressed by read-through transcription or that they can overlap the 5' end or even the entire gene. These results require a critical reevaluation of whether origins form at specific DNA elements and/or epigenetic signals or require no such determinants.
© 2015 Hyrien.
Figures
References
-
- Aladjem M.I., Rodewald L.W., Lin C.M., Bowman S., Cimbora D.M., Brody L.L., Epner E.M., Groudine M., and Wahl G.M.. 2002. Replication initiation patterns in the β-globin loci of totipotent and differentiated murine cells: evidence for multiple initiation regions. Mol. Cell. Biol. 22:442–452 10.1128/MCB.22.2.442-452.2002 - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
