

Amyloid β oligomers in Alzheimer's disease pathogenesis, treatment, and diagnosis
- PMID: 25604547
- PMCID: PMC4390393
- DOI: 10.1007/s00401-015-1386-3
Amyloid β oligomers in Alzheimer's disease pathogenesis, treatment, and diagnosis
Abstract
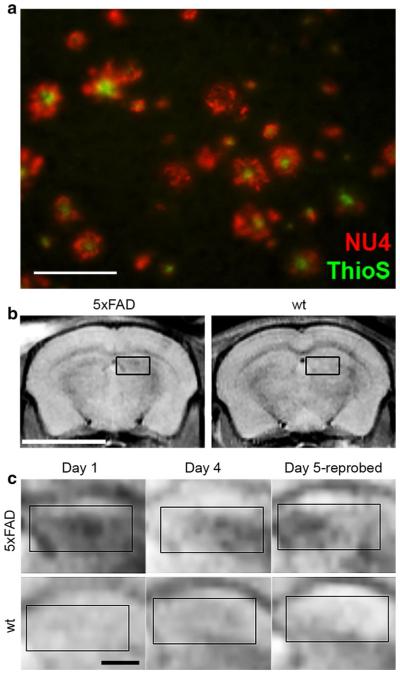
Protein aggregation is common to dozens of diseases including prionoses, diabetes, Parkinson's and Alzheimer's. Over the past 15 years, there has been a paradigm shift in understanding the structural basis for these proteinopathies. Precedent for this shift has come from investigation of soluble Aβ oligomers (AβOs), toxins now widely regarded as instigating neuron damage leading to Alzheimer's dementia. Toxic AβOs accumulate in AD brain and constitute long-lived alternatives to the disease-defining Aβ fibrils deposited in amyloid plaques. Key experiments using fibril-free AβO solutions demonstrated that while Aβ is essential for memory loss, the fibrillar Aβ in amyloid deposits is not the agent. The AD-like cellular pathologies induced by AβOs suggest their impact provides a unifying mechanism for AD pathogenesis, explaining why early stage disease is specific for memory and accounting for major facets of AD neuropathology. Alternative ideas for triggering mechanisms are being actively investigated. Some research favors insertion of AβOs into membrane, while other evidence supports ligand-like accumulation at particular synapses. Over a dozen candidate toxin receptors have been proposed. AβO binding triggers a redistribution of critical synaptic proteins and induces hyperactivity in metabotropic and ionotropic glutamate receptors. This leads to Ca(2+) overload and instigates major facets of AD neuropathology, including tau hyperphosphorylation, insulin resistance, oxidative stress, and synapse loss. Because different species of AβOs have been identified, a remaining question is which oligomer is the major pathogenic culprit. The possibility has been raised that more than one species plays a role. Despite some key unknowns, the clinical relevance of AβOs has been established, and new studies are beginning to point to co-morbidities such as diabetes and hypercholesterolemia as etiological factors. Because pathogenic AβOs appear early in the disease, they offer appealing targets for therapeutics and diagnostics. Promising therapeutic strategies include use of CNS insulin signaling enhancers to protect against the presence of toxins and elimination of the toxins through use of highly specific AβO antibodies. An AD-dependent accumulation of AβOs in CSF suggests their potential use as biomarkers and new AβO probes are opening the door to brain imaging. Overall, current evidence indicates that Aβ oligomers provide a substantive molecular basis for the cause, treatment and diagnosis of Alzheimer's disease.
Figures

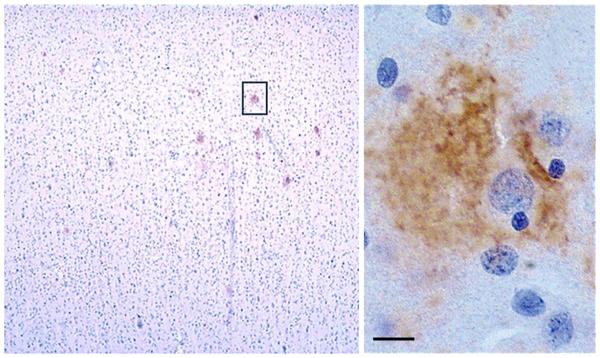

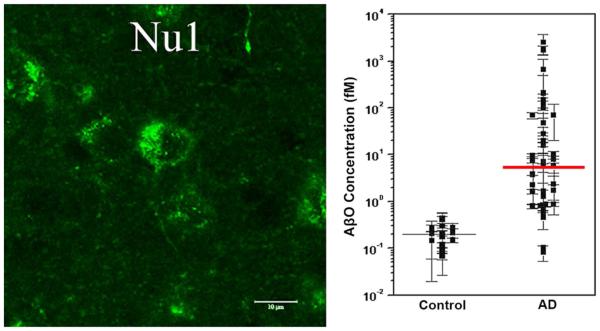

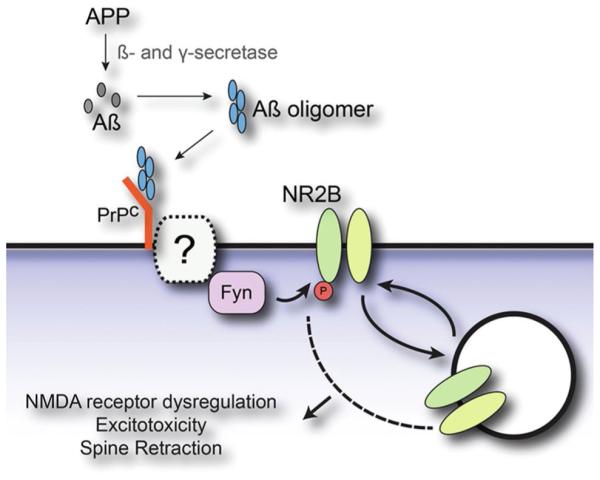

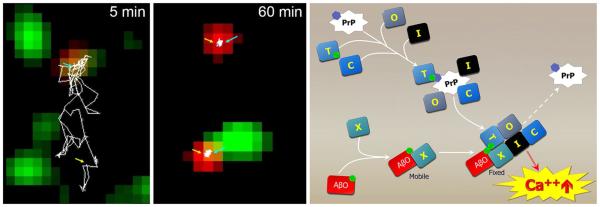



References
-
- Adolfsson O, Pihlgren M, Toni N, Varisco Y, Buccarello AL, Antoniello K, Lohmann S, Piorkowska K, Gafner V, Atwal JK, Maloney J, Chen M, Gogineni A, Weimer RM, Mortensen DL, Friesenhahn M, Ho C, Paul R, Pfeifer A, Muhs A, Watts RJ. An effector-reduced anti-beta-amyloid (Abeta) antibody with unique abeta binding properties promotes neuroprotection and glial engulfment of Abeta. J Neurosci Off J Soc Neurosci. 2012;32(28):9677–9689. doi:10.1523/jneurosci.4742-11.2012. - PMC - PubMed
-
- Arispe N. Architecture of the Alzheimer’s A beta P ion channel pore. J Membr Biol. 2004;197(1):33–48. doi:10.1007/ s00232-003-0638-7. - PubMed
-
- Bartl J, Meyer A, Brendler S, Riederer P, Grunblatt E. Different effects of soluble and aggregated amyloid beta42 on gene/protein expression and enzyme activity involved in insulin and APP pathways. J Neural Trans Vienna Austria 1996. 2013;120(1):113–120. doi:10.1007/s00702-012-0852-5. - PubMed
-
- Benilova I, Karran E, De Strooper B. The toxic Abeta oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat Neurosci. 2012;15(3):349–357. doi:10.1038/nn.3028. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
