

RMND1 deficiency associated with neonatal lactic acidosis, infantile onset renal failure, deafness, and multiorgan involvement
- PMID: 25604853
- PMCID: PMC4592087
- DOI: 10.1038/ejhg.2014.293
RMND1 deficiency associated with neonatal lactic acidosis, infantile onset renal failure, deafness, and multiorgan involvement
Abstract
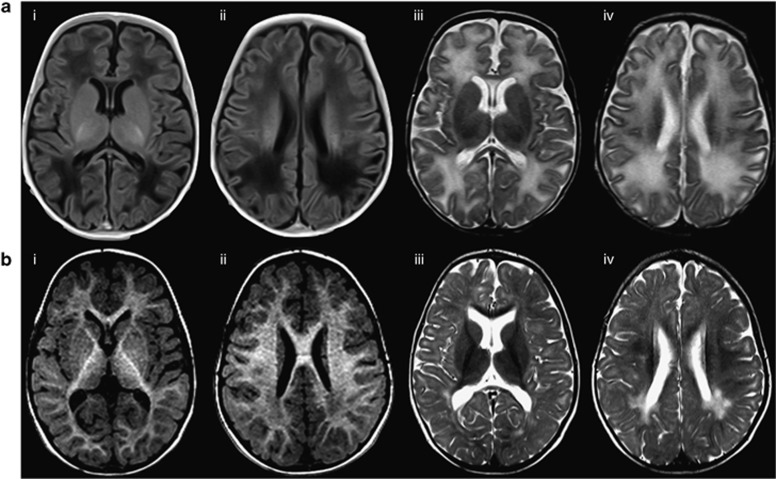
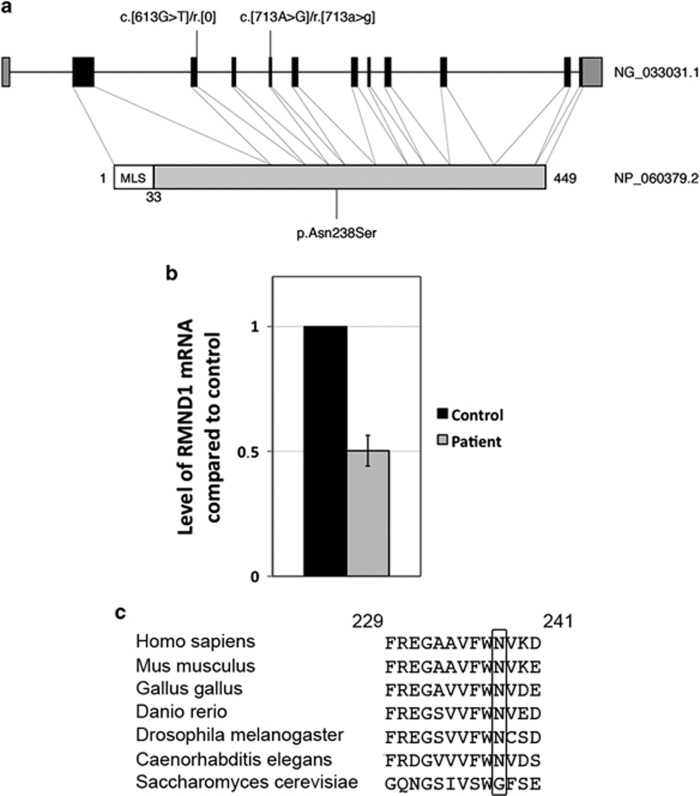
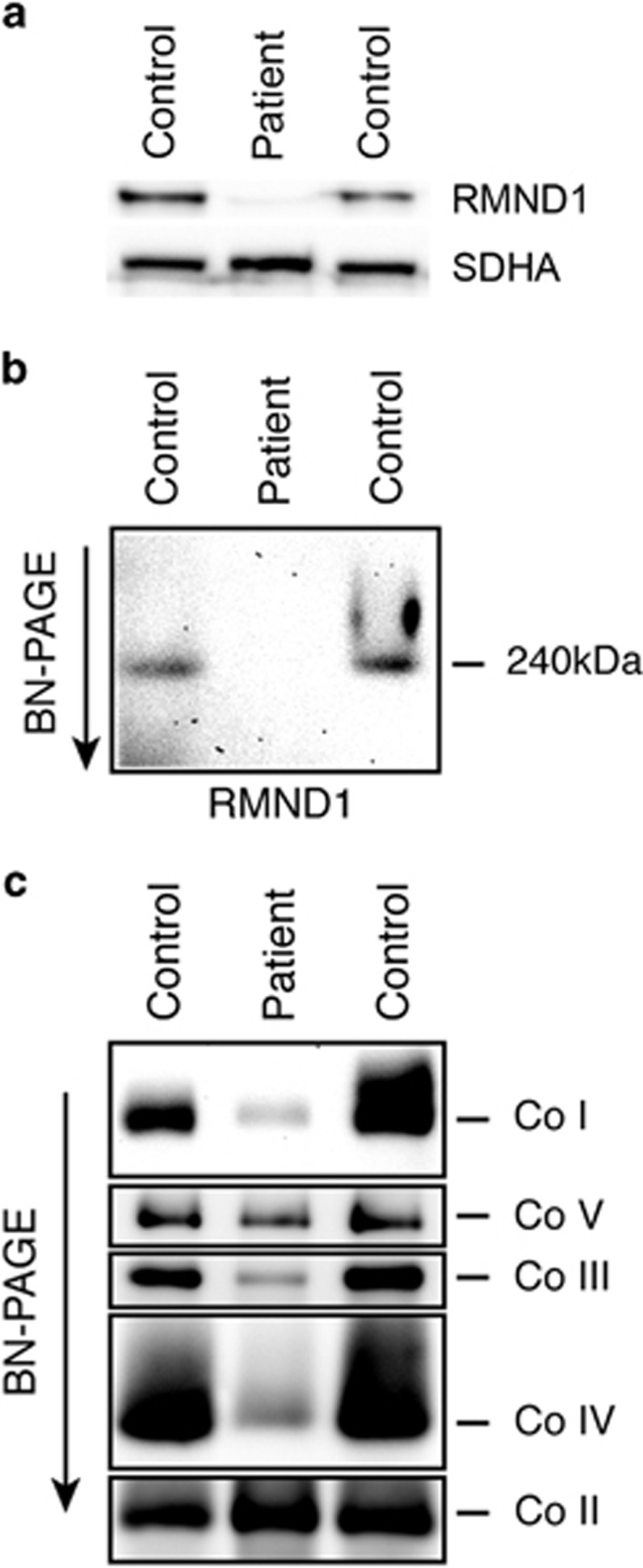
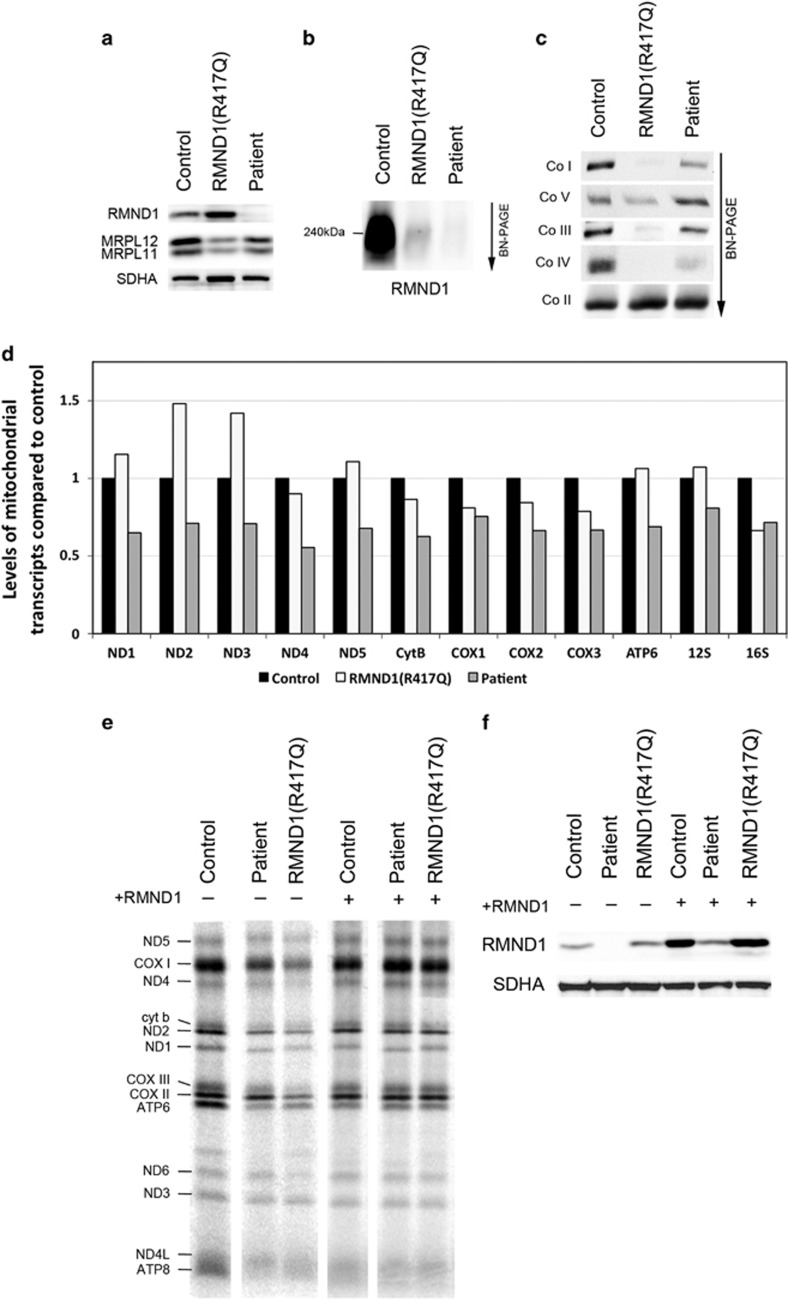
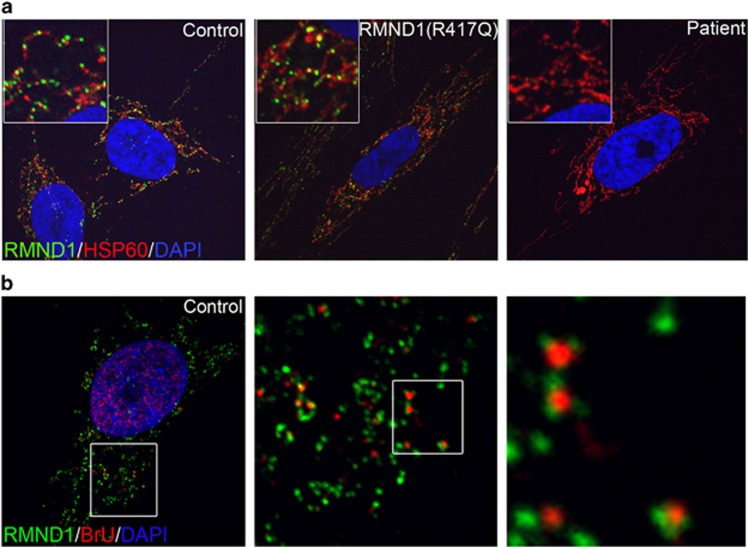
RMND1 is an integral inner membrane mitochondrial protein that assembles into a large 240 kDa complex to support translation of the 13 polypeptides encoded on mtDNA, all of which are essential subunits of the oxidative phosphorylation (OXPHOS) complexes. Variants in RMND1 produce global defects in mitochondrial translation and were first reported in patients with severe neurological phenotypes leading to mortality in the first months of life. Using whole-exome sequencing, we identified compound heterozygous RMND1 variants in a 4-year-old patient with congenital lactic acidosis, severe myopathy, hearing loss, renal failure, and dysautonomia. The levels of mitochondrial ribosome proteins were reduced in patient fibroblasts, causing a translation defect, which was rescued by expression of the wild-type cDNA. RMND1 was almost undetectable by immunoblot analysis in patient muscle and fibroblasts. BN-PAGE analysis showed a severe combined OXPHOS assembly defect that was more prominent in patient muscle than in fibroblasts. Immunofluorescence experiments showed that RMND1 localizes to discrete foci in the mitochondrial network, juxtaposed to RNA granules where the primary mitochondrial transcripts are processed. RMND1 foci were not detected in patient fibroblasts. We hypothesize that RMND1 acts to anchor or stabilize the mitochondrial ribosome near the sites where the mRNAs are matured, spatially coupling post-transcriptional handling mRNAs with their translation, and that loss of function variants in RMND1 are associated with a unique constellation of clinical phenotypes that vary with the severity of the mitochondrial translation defect.
Figures
References
-
- 1Coenen MJ, Antonicka H, Ugalde C et al: Mutant mitochondrial elongation factor G1 and combined oxidative phosphorylation deficiency. N Engl J Med 2004; 351: 2080–2086. - PubMed
-
- 2Hallberg BM, Larsson NG: Making proteins in the powerhouse. Cell Metab 2014; 20: 226–240. - PubMed
-
- 3Weraarpachai W, Antonicka H, Sasarman F et al: Mutation in TACO1, encoding a translational activator of COX I, results in cytochrome c oxidase deficiency and late-onset Leigh syndrome. Nat Genet 2009; 41: 833–837. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
