

Microtubule-targeting agents augment the toxicity of DNA-damaging agents by disrupting intracellular trafficking of DNA repair proteins
- PMID: 25605897
- PMCID: PMC4321245
- DOI: 10.1073/pnas.1416418112
Microtubule-targeting agents augment the toxicity of DNA-damaging agents by disrupting intracellular trafficking of DNA repair proteins
Abstract
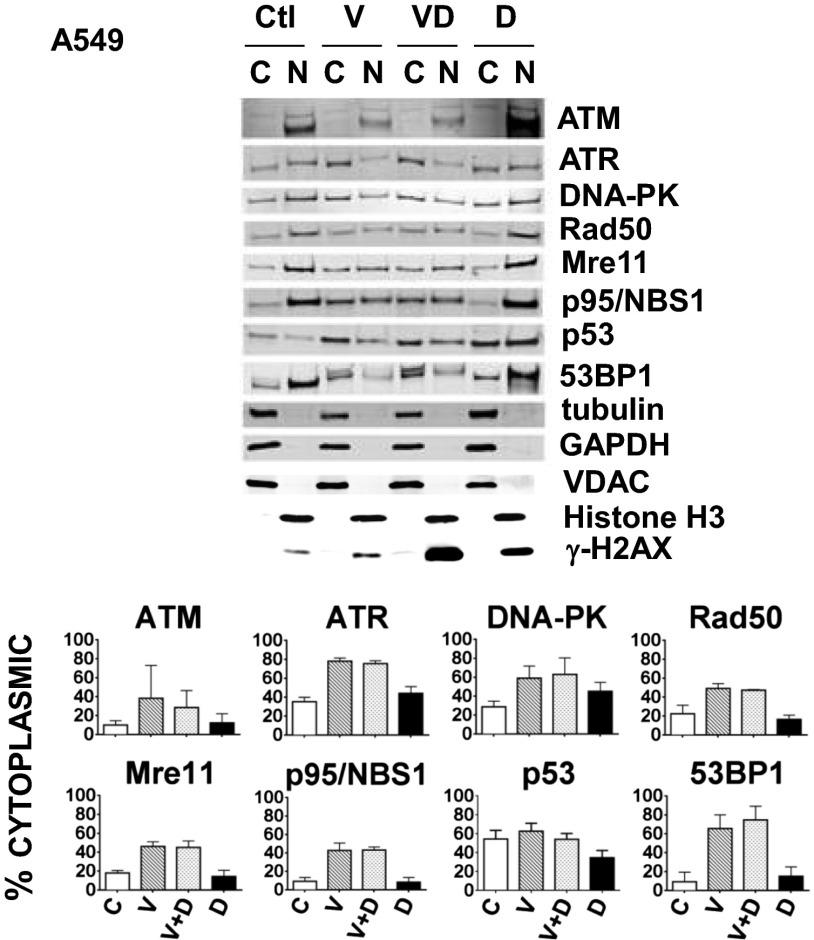
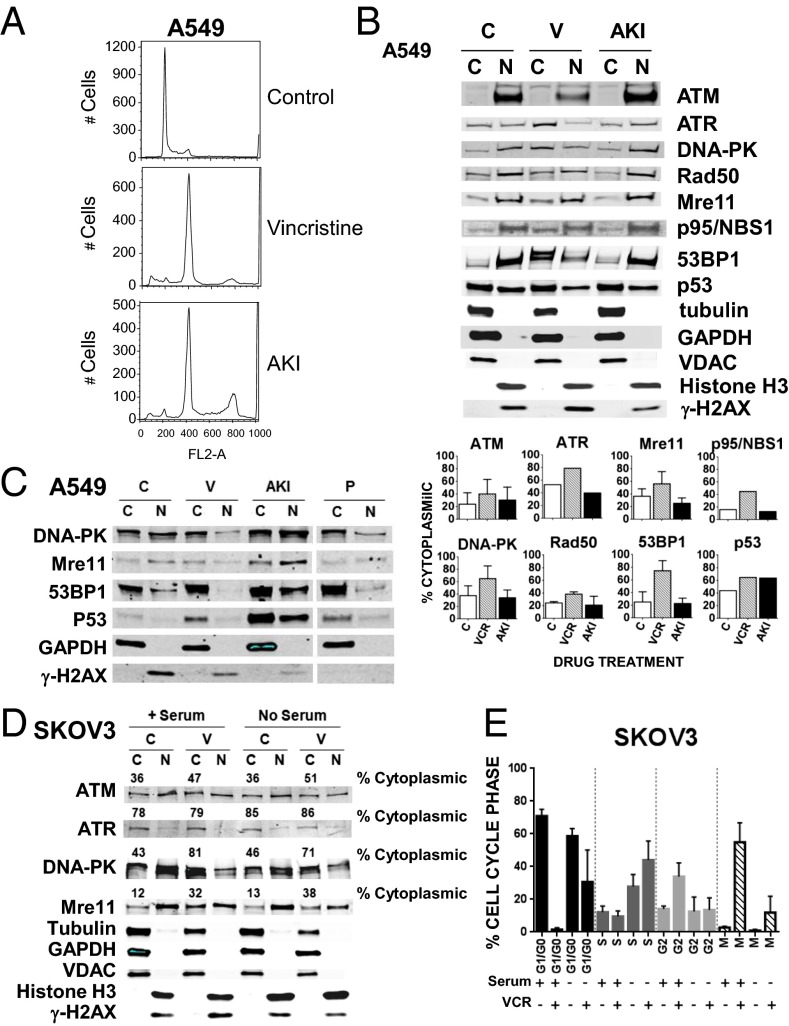
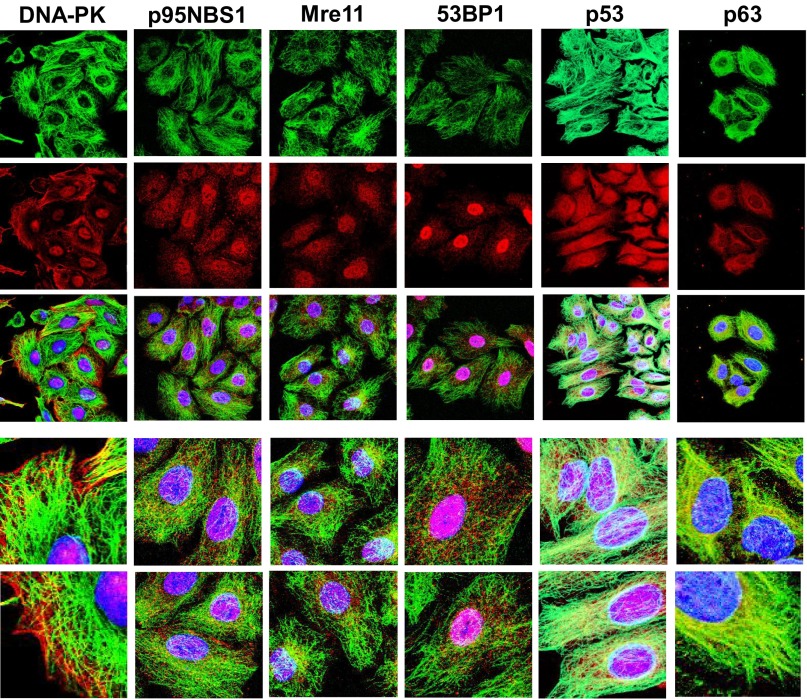
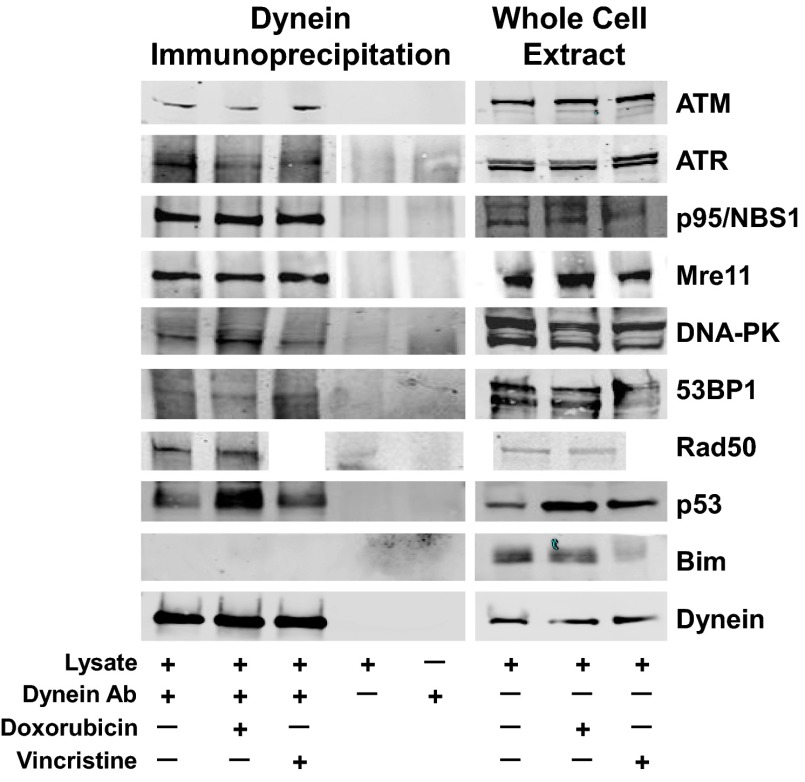
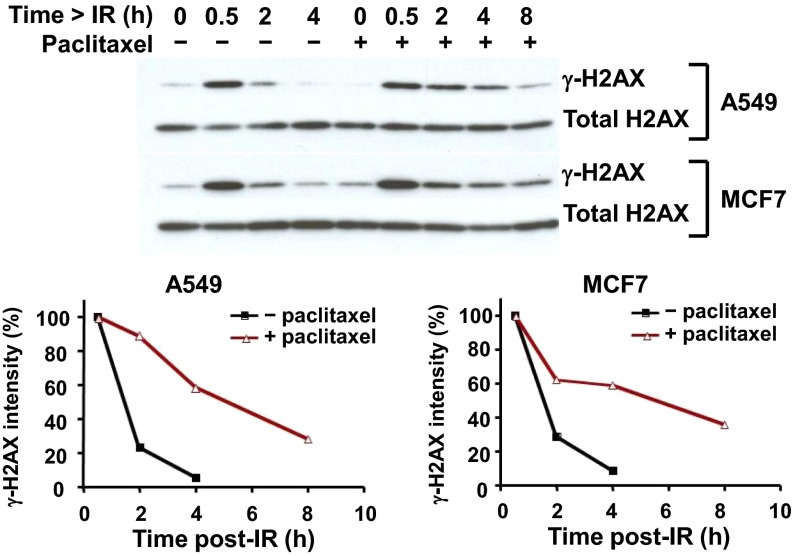
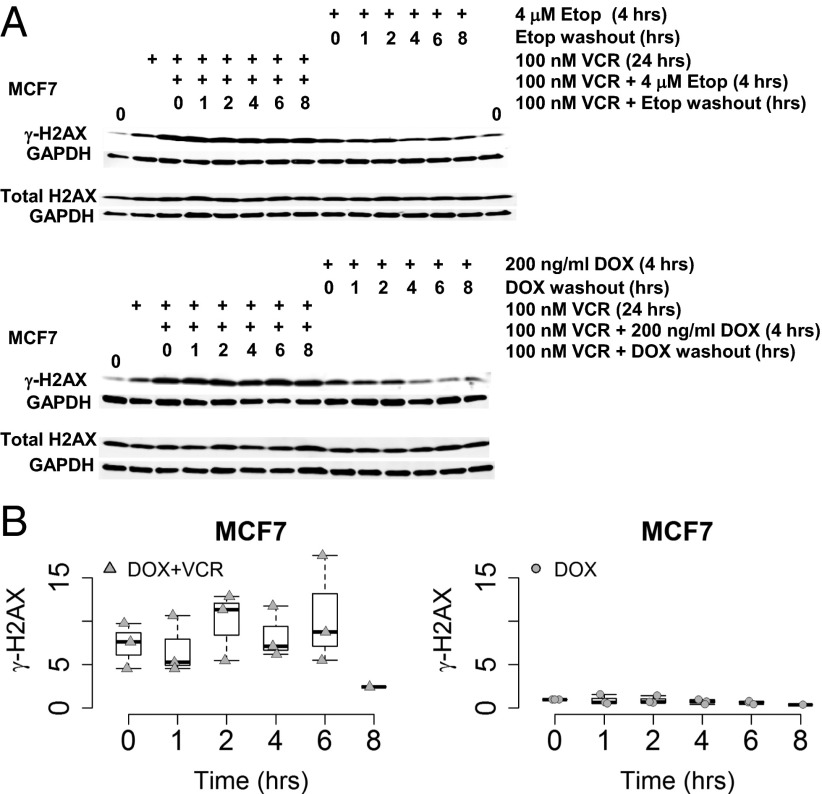
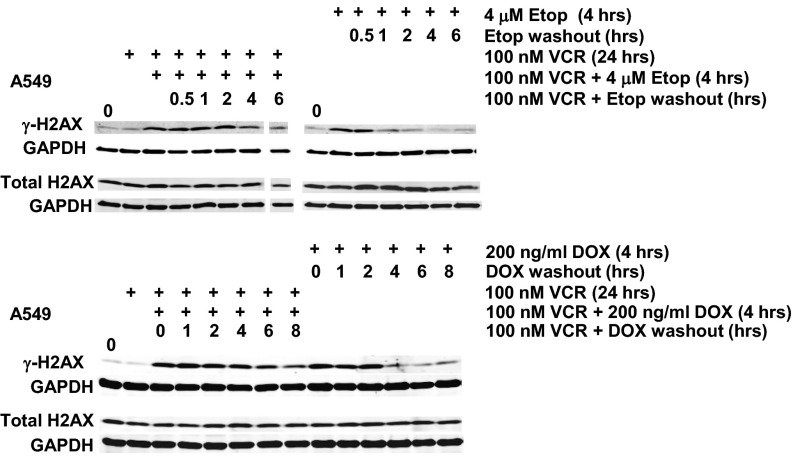
The paradigm that microtubule-targeting agents (MTAs) cause cell death via mitotic arrest applies to rapidly dividing cells but cannot explain MTA activity in slowly growing human cancers. Many preferred cancer regimens combine a MTA with a DNA-damaging agent (DDA). We hypothesized that MTAs synergize with DDAs by interfering with trafficking of DNA repair proteins on interphase microtubules. We investigated nine proteins involved in DNA repair: ATM, ATR, DNA-PK, Rad50, Mre11, p95/NBS1, p53, 53BP1, and p63. The proteins were sequestered in the cytoplasm by vincristine and paclitaxel but not by an aurora kinase inhibitor, colocalized with tubulin by confocal microscopy and coimmunoprecipitated with the microtubule motor dynein. Furthermore, adding MTAs to radiation, doxorubicin, or etoposide led to more sustained γ-H2AX levels. We conclude DNA damage-repair proteins traffic on microtubules and addition of MTAs sequesters them in the cytoplasm, explaining why MTA/DDA combinations are common anticancer regimens.
Keywords: DNA repair protein trafficking; DNA-damaging agents; combination chemotherapy; microtubule targeting agents; targeted therapies.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Milas L, et al. Kinetics of mitotic arrest and apoptosis in murine mammary and ovarian tumors treated with taxol. Cancer Chemother Pharmacol. 1995;35(4):297–303. - PubMed
-
- Horton JK, Houghton PJ, Houghton JA. Relationships between tumor responsiveness, vincristine pharmacokinetics and arrest of mitosis in human tumor xenografts. Biochem Pharmacol. 1988;37(20):3995–4000. - PubMed
-
- Komlodi-Pasztor E, Sackett D, Wilkerson J, Fojo T. Mitosis is not a key target of microtubule agents in patient tumors. Nat Rev Clin Oncol. 2011;8(4):244–250. - PubMed
-
- Komlodi-Pasztor E, Sackett DL, Fojo AT. Inhibitors targeting mitosis: Tales of how great drugs against a promising target were brought down by a flawed rationale. Clin Cancer Res. 2012;18(1):51–63. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
