

Structure of the key species in the enzymatic oxidation of methane to methanol
- PMID: 25607364
- PMCID: PMC4429310
- DOI: 10.1038/nature14160
Structure of the key species in the enzymatic oxidation of methane to methanol
Abstract
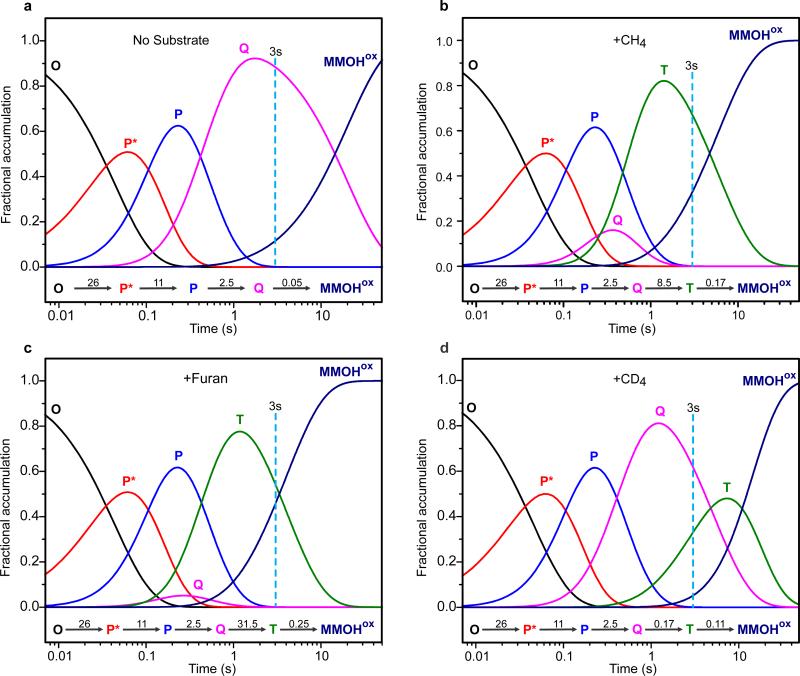
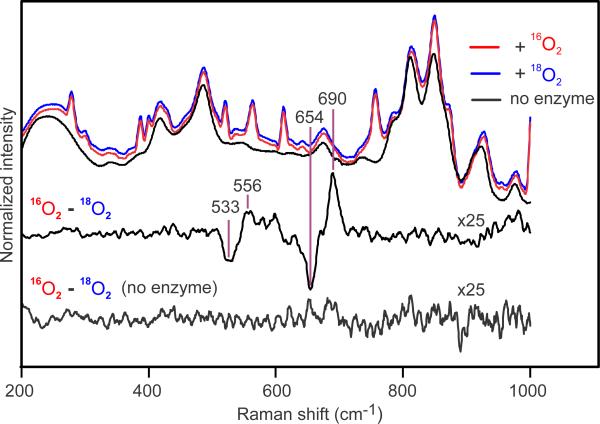
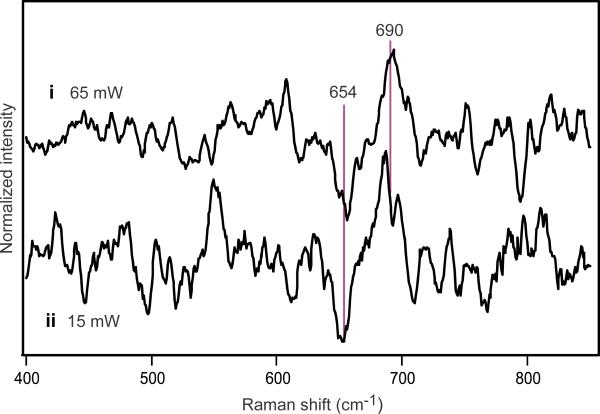
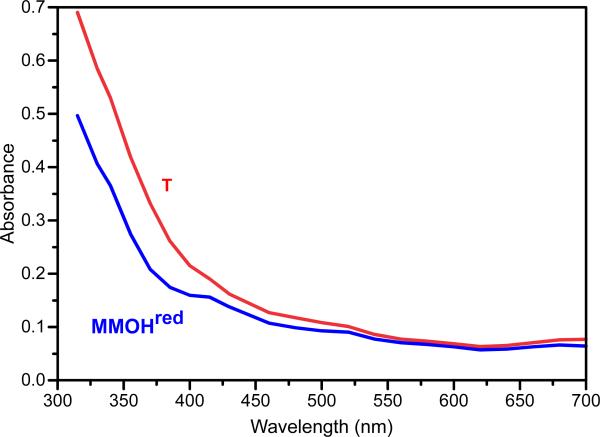
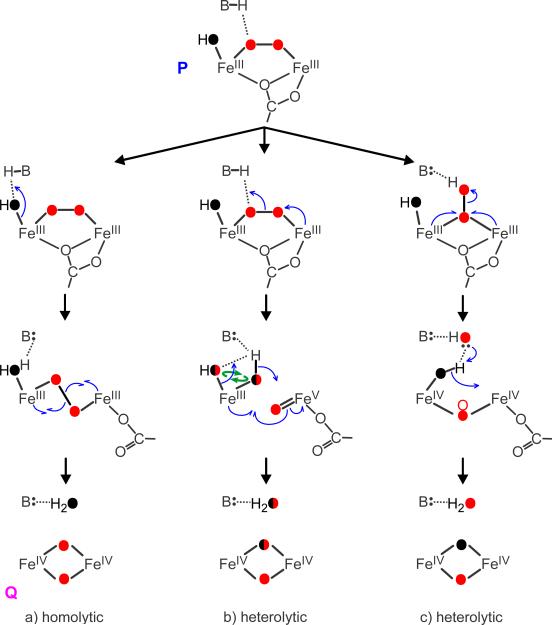
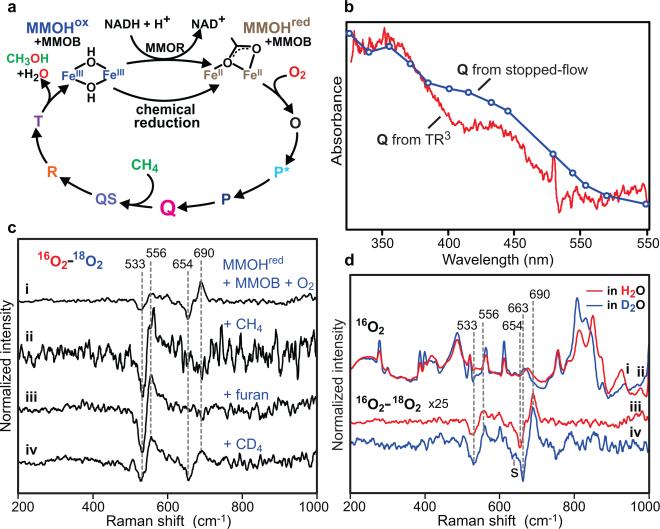
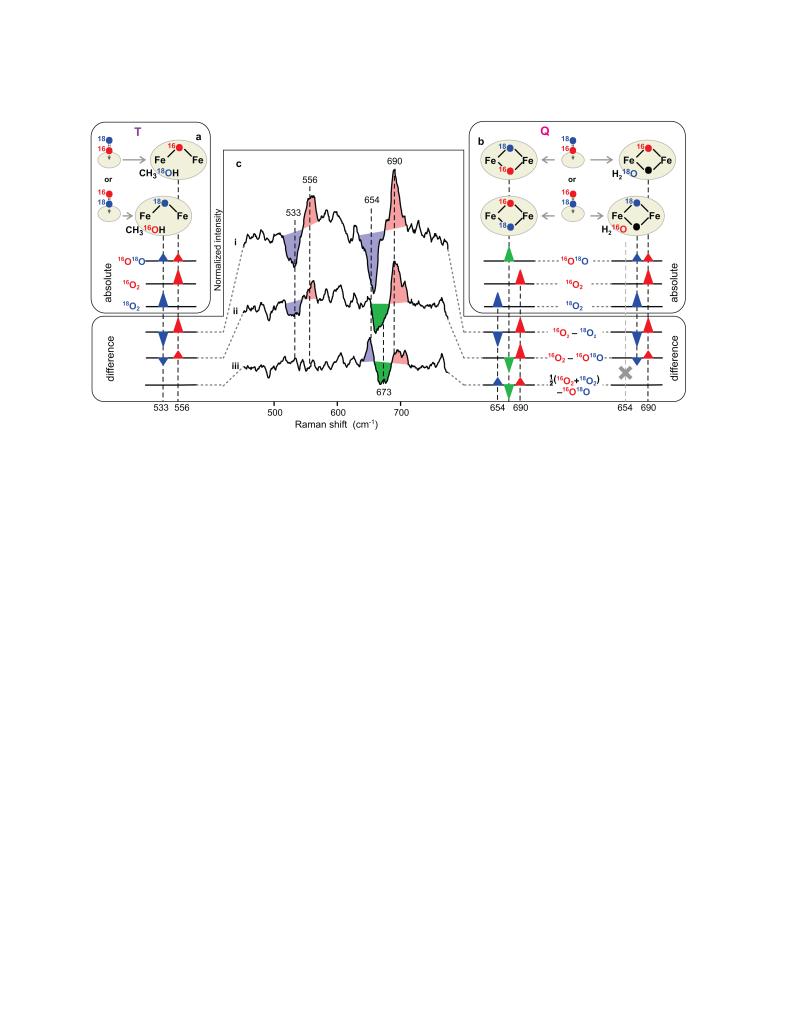
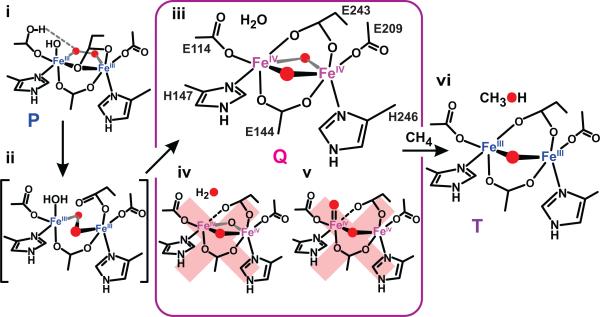
Methane monooxygenase (MMO) catalyses the O2-dependent conversion of methane to methanol in methanotrophic bacteria, thereby preventing the atmospheric egress of approximately one billion tons of this potent greenhouse gas annually. The key reaction cycle intermediate of the soluble form of MMO (sMMO) is termed compound Q (Q). Q contains a unique dinuclear Fe(IV) cluster that reacts with methane to break an exceptionally strong 105 kcal mol(-1) C-H bond and insert one oxygen atom. No other biological oxidant, except that found in the particulate form of MMO, is capable of such catalysis. The structure of Q remains controversial despite numerous spectroscopic, computational and synthetic model studies. A definitive structural assignment can be made from resonance Raman vibrational spectroscopy but, despite efforts over the past two decades, no vibrational spectrum of Q has yet been obtained. Here we report the core structures of Q and the following product complex, compound T, using time-resolved resonance Raman spectroscopy (TR(3)). TR(3) permits fingerprinting of intermediates by their unique vibrational signatures through extended signal averaging for short-lived species. We report unambiguous evidence that Q possesses a bis-μ-oxo diamond core structure and show that both bridging oxygens originate from O2. This observation strongly supports a homolytic mechanism for O-O bond cleavage. We also show that T retains a single oxygen atom from O2 as a bridging ligand, while the other oxygen atom is incorporated into the product. Capture of the extreme oxidizing potential of Q is of great contemporary interest for bioremediation and the development of synthetic approaches to methane-based alternative fuels and chemical industry feedstocks. Insight into the formation and reactivity of Q from the structure reported here is an important step towards harnessing this potential.
Figures
Comment in
-
Biochemistry: Breaking methane.Nature. 2015 Feb 19;518(7539):309-10. doi: 10.1038/nature14199. Epub 2015 Jan 21. Nature. 2015. PMID: 25607367 Free PMC article.
Similar articles
-
Nuclear Resonance Vibrational Spectroscopic Definition of the Fe(IV)2 Intermediate Q in Methane Monooxygenase and Its Reactivity.J Am Chem Soc. 2021 Oct 6;143(39):16007-16029. doi: 10.1021/jacs.1c05436. Epub 2021 Sep 27. J Am Chem Soc. 2021. PMID: 34570980 Free PMC article.
-
A diiron(IV) complex that cleaves strong C-H and O-H bonds.Nat Chem. 2009 May;1(2):145-50. doi: 10.1038/nchem.162. Nat Chem. 2009. PMID: 19885382 Free PMC article.
-
Dioxygen activation in soluble methane monooxygenase.Acc Chem Res. 2011 Apr 19;44(4):280-8. doi: 10.1021/ar1001473. Epub 2011 Mar 10. Acc Chem Res. 2011. PMID: 21391602 Free PMC article. Review.
-
Regulation of methane monooxygenase catalysis based on size exclusion and quantum tunneling.Biochemistry. 2006 Feb 14;45(6):1685-92. doi: 10.1021/bi051605g. Biochemistry. 2006. PMID: 16460015
-
Direct Methane Oxidation by Copper- and Iron-Dependent Methane Monooxygenases.Chem Rev. 2024 Feb 14;124(3):1288-1320. doi: 10.1021/acs.chemrev.3c00727. Epub 2024 Feb 2. Chem Rev. 2024. PMID: 38305159 Free PMC article. Review.
Cited by
-
Small-Molecule Tunnels in Metalloenzymes Viewed as Extensions of the Active Site.Acc Chem Res. 2021 May 4;54(9):2185-2195. doi: 10.1021/acs.accounts.1c00058. Epub 2021 Apr 22. Acc Chem Res. 2021. PMID: 33886257 Free PMC article. Review.
-
Toward the synthesis of more reactive S = 2 non-heme oxoiron(IV) complexes.Acc Chem Res. 2015 Aug 18;48(8):2443-52. doi: 10.1021/acs.accounts.5b00244. Epub 2015 Jul 15. Acc Chem Res. 2015. PMID: 26176555 Free PMC article.
-
Sizing up a supercharged ferryl.Proc Natl Acad Sci U S A. 2018 May 1;115(18):4532-4534. doi: 10.1073/pnas.1804490115. Epub 2018 Apr 16. Proc Natl Acad Sci U S A. 2018. PMID: 29666275 Free PMC article. No abstract available.
-
Methane: Fuel or Exhaust at the Emergence of Life?Astrobiology. 2017 Oct;17(10):1053-1066. doi: 10.1089/ast.2016.1599. Epub 2017 Sep 26. Astrobiology. 2017. PMID: 28949766 Free PMC article.
-
Crystal structure of CmlI, the arylamine oxygenase from the chloramphenicol biosynthetic pathway.J Biol Inorg Chem. 2016 Sep;21(5-6):589-603. doi: 10.1007/s00775-016-1363-x. Epub 2016 May 26. J Biol Inorg Chem. 2016. PMID: 27229511 Free PMC article.
References
-
- Lee SK, Nesheim JC, Lipscomb JD. Transient intermediates of the methane monooxygenase catalytic cycle. J. Biol. Chem. 1993;268:21569–21577. - PubMed
-
- Lee SK, Fox BG, Froland WA, Lipscomb JD, Münck E. A transient intermediate of the methane monooxygenase catalytic cycle containing a FeIVFeIV cluster. J. Am. Chem. Soc. 1993;115:6450–6451.
-
- Shu L, et al. An FeIV2O2 diamond core structure for the key intermediate Q of methane monooxygenase. Science. 1997;275:515–518. - PubMed
-
- Dunietz BD, et al. Large scale ab initio quantum chemical calculation of the intermediates in the soluble methane monooxygenase catalytic cycle. J. Am. Chem. Soc. 2000;122:2828–2839.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
