

Enhancer sequence variants and transcription-factor deregulation synergize to construct pathogenic regulatory circuits in B-cell lymphoma
- PMID: 25607463
- PMCID: PMC4302272
- DOI: 10.1016/j.immuni.2014.12.021
Enhancer sequence variants and transcription-factor deregulation synergize to construct pathogenic regulatory circuits in B-cell lymphoma
Abstract
Most B-cell lymphomas arise in the germinal center (GC), where humoral immune responses evolve from potentially oncogenic cycles of mutation, proliferation, and clonal selection. Although lymphoma gene expression diverges significantly from GC B cells, underlying mechanisms that alter the activities of corresponding regulatory elements (REs) remain elusive. Here we define the complete pathogenic circuitry of human follicular lymphoma (FL), which activates or decommissions REs from normal GC B cells and commandeers enhancers from other lineages. Moreover, independent sets of transcription factors, whose expression was deregulated in FL, targeted commandeered versus decommissioned REs. Our approach revealed two distinct subtypes of low-grade FL, whose pathogenic circuitries resembled GC B or activated B cells. FL-altered enhancers also were enriched for sequence variants, including somatic mutations, which disrupt transcription-factor binding and expression of circuit-linked genes. Thus, the pathogenic regulatory circuitry of FL reveals distinct genetic and epigenetic etiologies for GC B-cell transformation.
Copyright © 2015 Elsevier Inc. All rights reserved.
Figures
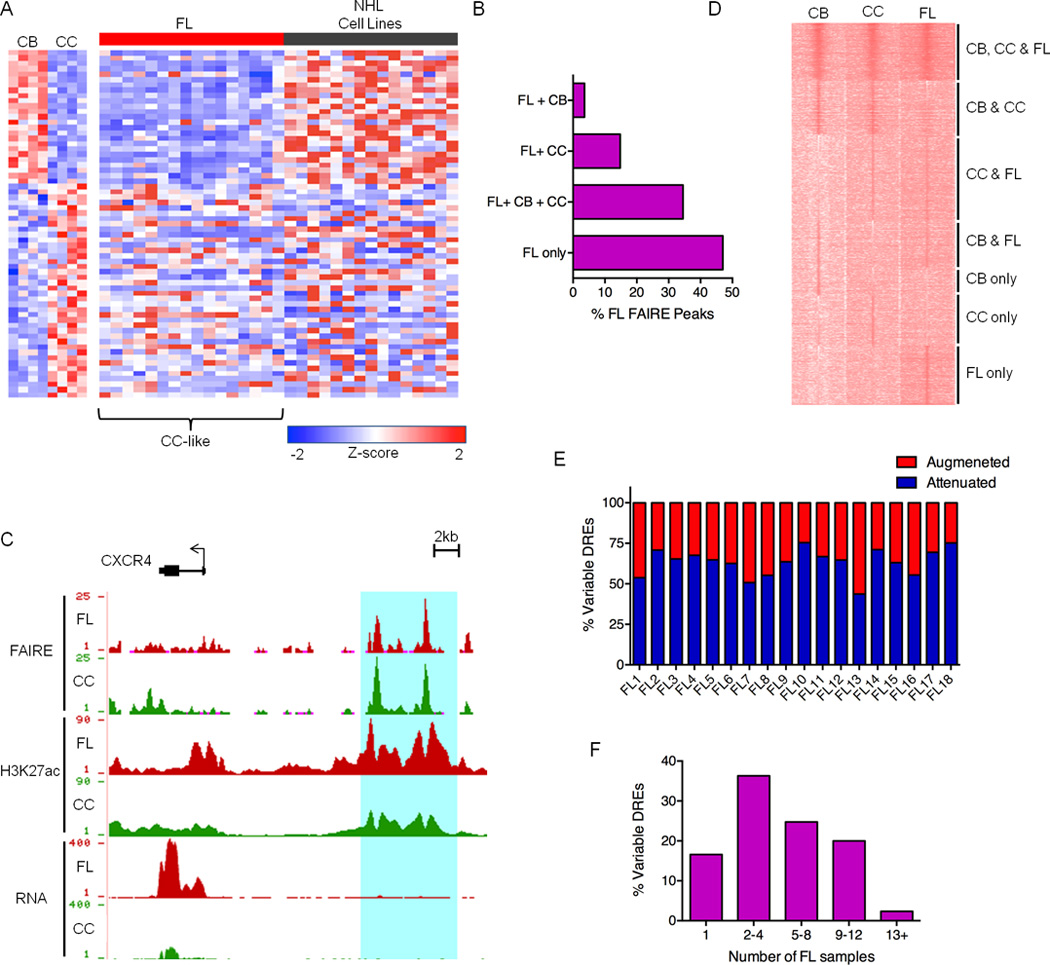
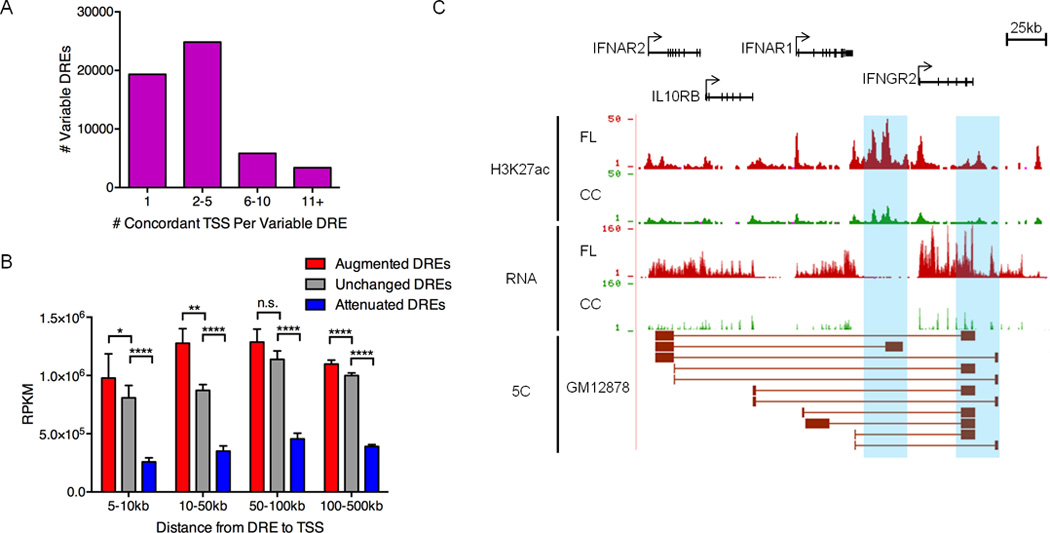
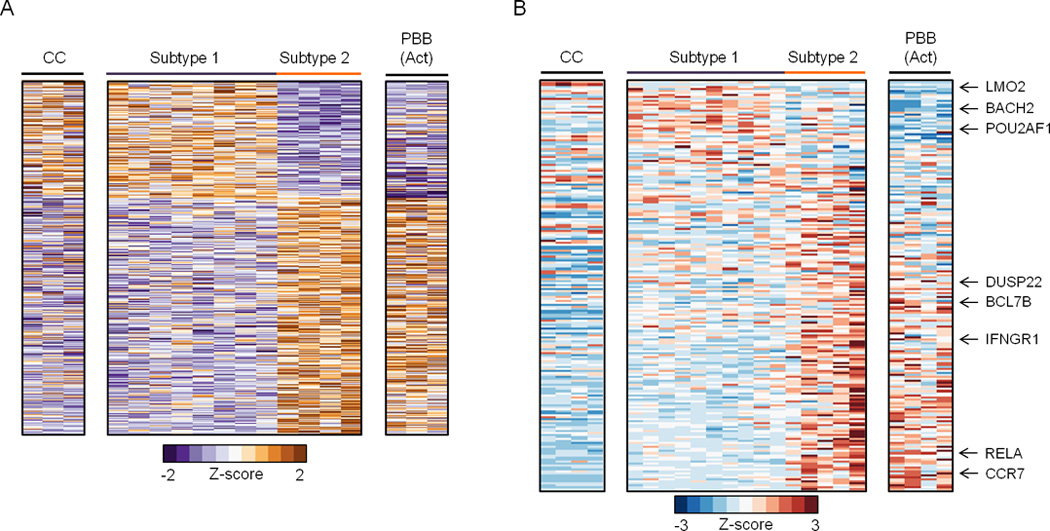
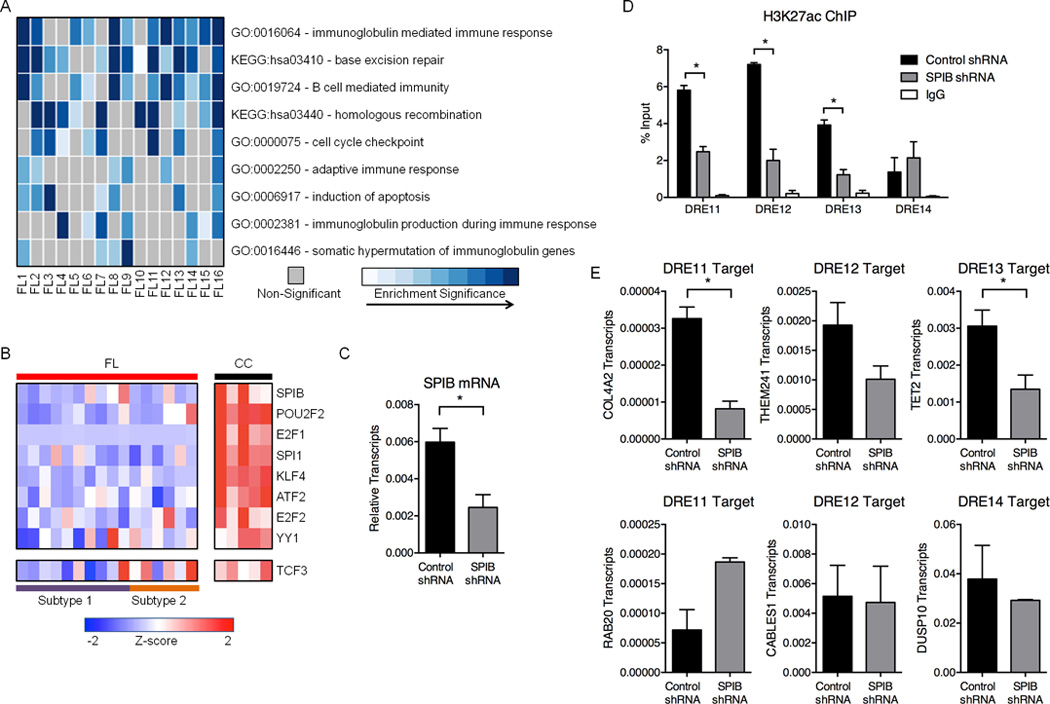
References
-
- Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–511. - PubMed
-
- Argiropoulos B, Humphries RK. Hox genes in hematopoiesis and leukemogenesis. Oncogene. 2007;26:6766–6776. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
