

Sequencing of first-strand cDNA library reveals full-length transcriptomes
- PMID: 25607527
- PMCID: PMC5054741
- DOI: 10.1038/ncomms7002
Sequencing of first-strand cDNA library reveals full-length transcriptomes
Abstract
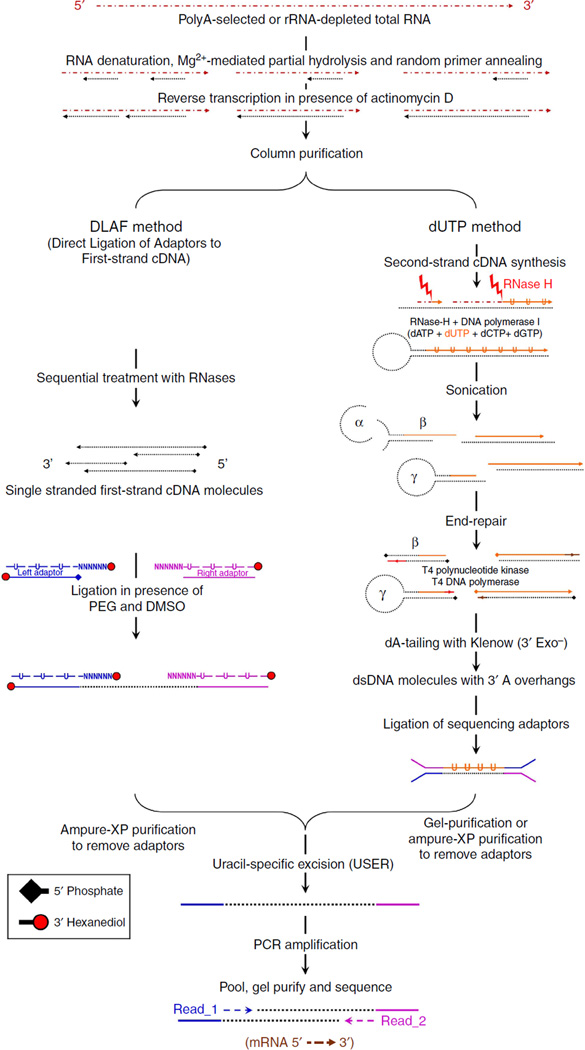
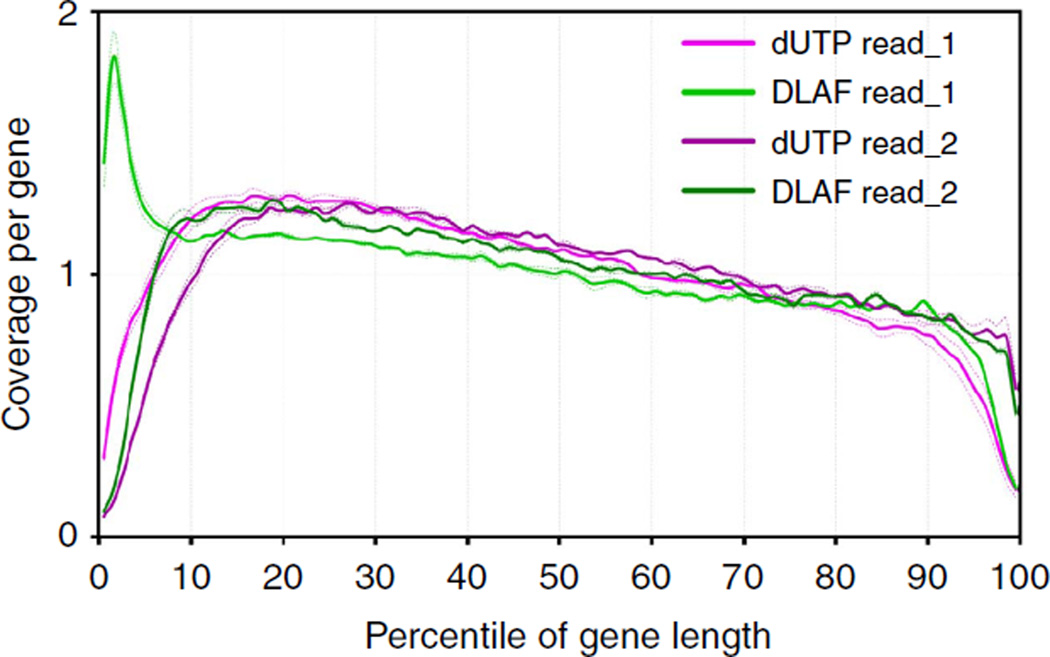
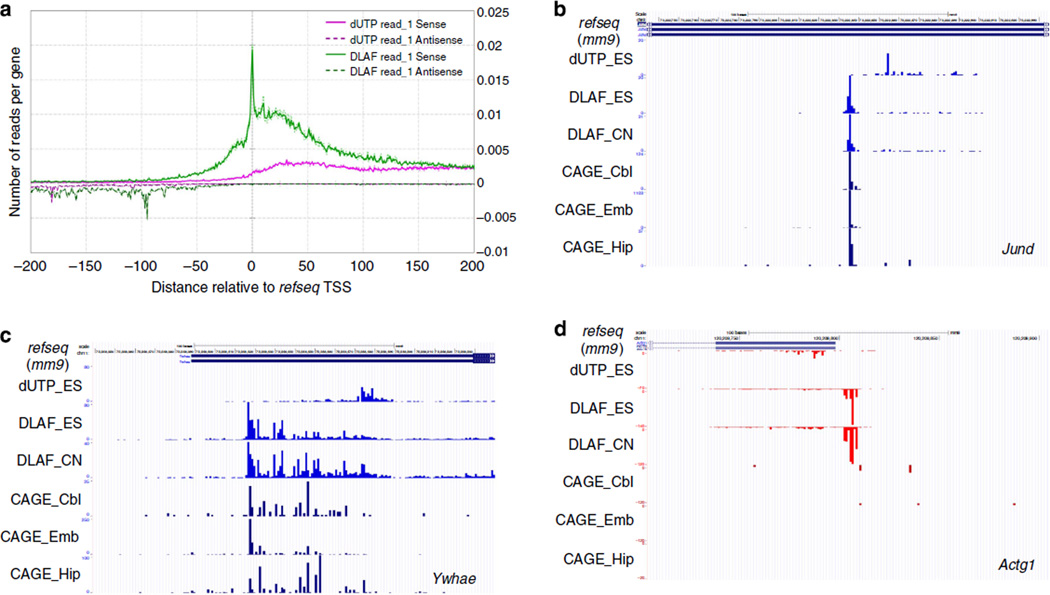
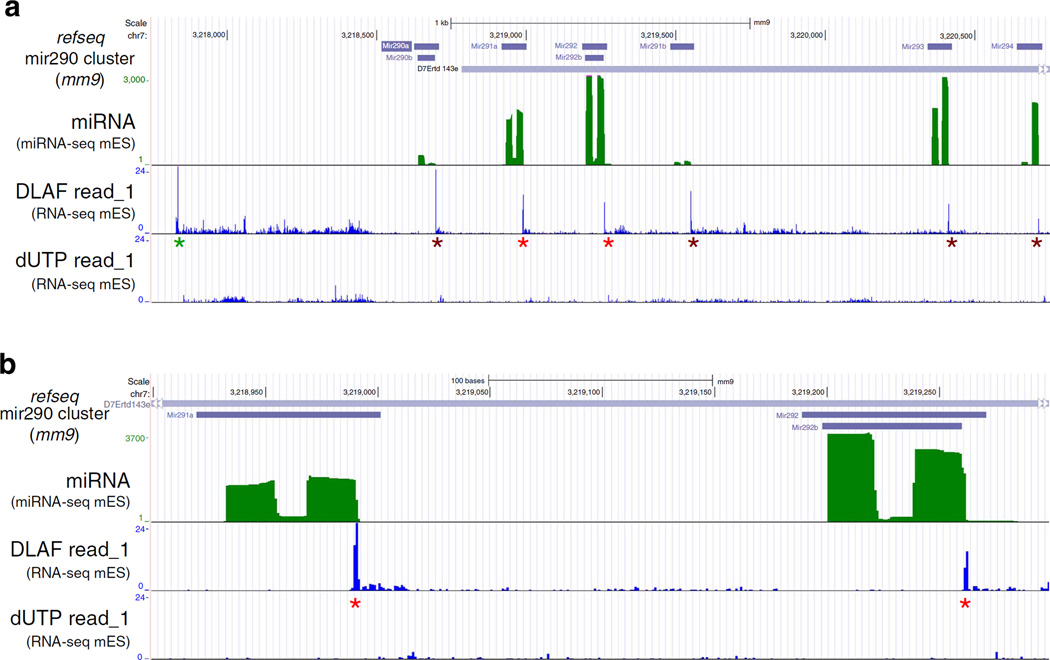
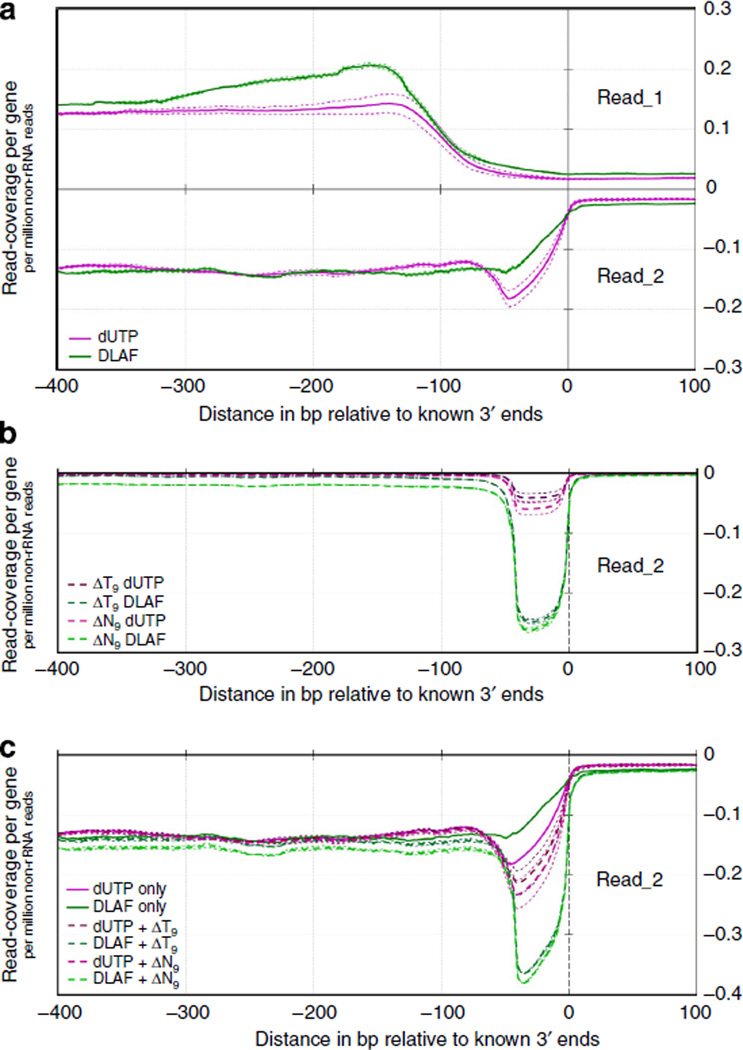
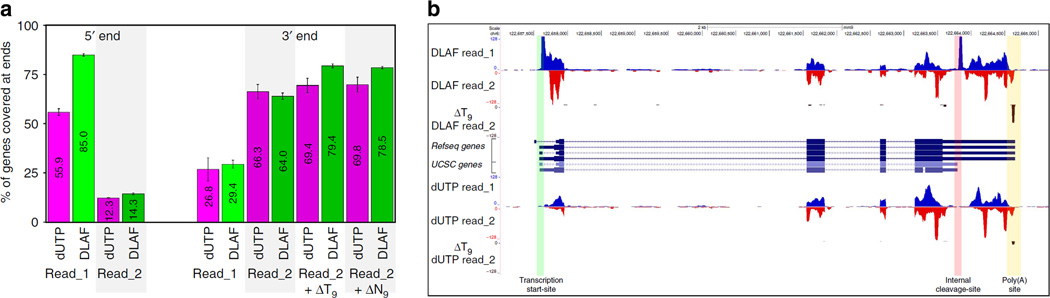
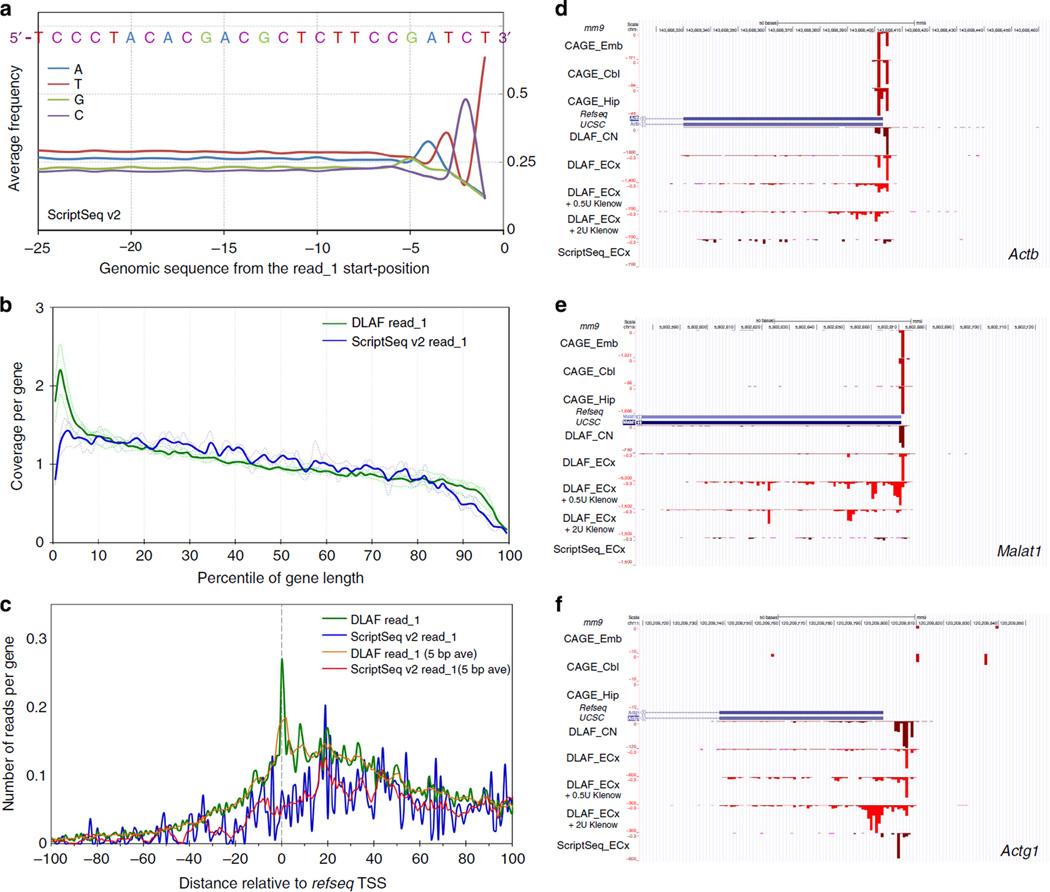
Massively parallel strand-specific sequencing of RNA (ssRNA-seq) has emerged as a powerful tool for profiling complex transcriptomes. However, many current methods for ssRNA-seq suffer from the underrepresentation of both the 5' and 3' ends of RNAs, which can be attributed to second-strand cDNA synthesis. The 5' and 3' ends of RNA harbour crucial information for gene regulation; namely, transcription start sites (TSSs) and polyadenylation sites. Here we report a novel ssRNA-seq method that does not involve second-strand cDNA synthesis, as we Directly Ligate sequencing Adaptors to the First-strand cDNA (DLAF). This novel method with fewer enzymatic reactions results in a higher quality of the libraries than the conventional method. Sequencing of DLAF libraries followed by a novel analysis pipeline enables the profiling of both 5' ends and polyadenylation sites at near-base resolution. Therefore, DLAF offers the first genomics tool to obtain the 'full-length' transcriptome with a single library.
Figures
References
-
- Brenner S, et al. Gene expression analysis by massively parallel signature sequencing (MPSS) on microbead arrays. Nature Biotechnol. 2000;18:630–634. - PubMed
-
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 2008;5:621–628. - PubMed
-
- Zhu YY, Machleder EM, Chenchik A, Li R, Siebert PD. Reverse transcriptase template switching: a SMART approach for full-length cDNA library construction. BioTechniques. 2001;30:892–897. - PubMed
-
- Armour CD, et al. Digital transcriptome profiling using selective hexamer priming for cDNA synthesis. Nat. Methods. 2009;6:647–649. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
