

Mapping interactions between complement C3 and regulators using mutations in atypical hemolytic uremic syndrome
- PMID: 25608561
- PMCID: PMC4392009
- DOI: 10.1182/blood-2014-10-609073
Mapping interactions between complement C3 and regulators using mutations in atypical hemolytic uremic syndrome
Abstract
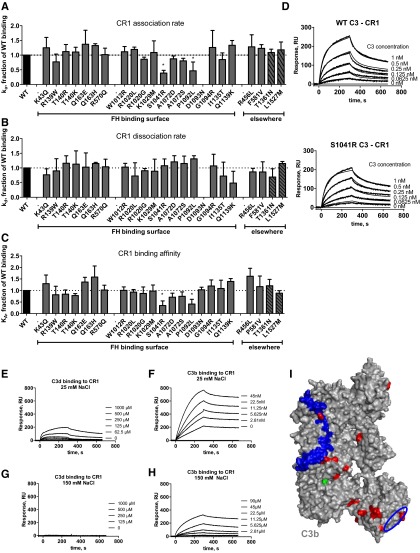
The pathogenesis of atypical hemolytic uremic syndrome (aHUS) is strongly linked to dysregulation of the alternative pathway of the complement system. Mutations in complement genes have been identified in about two-thirds of cases, with 5% to 15% being in C3. In this study, 23 aHUS-associated genetic changes in C3 were characterized relative to their interaction with the control proteins factor H (FH), membrane cofactor protein (MCP; CD46), and complement receptor 1 (CR1; CD35). In surface plasmon resonance experiments, 17 mutant recombinant proteins demonstrated a defect in binding to FH and/or MCP, whereas 2 demonstrated reduced binding to CR1. In the majority of cases, decreased binding affinity translated to a decrease in proteolytic inactivation (known as cofactor activity) of C3b via FH and MCP. These results were used to map the putative binding regions of C3b involved in the interaction with MCP and CR1 and interrogated relative to known FH binding sites. Seventy-six percent of patients with C3 mutations had low C3 levels that correlated with disease severity. This study expands our knowledge of the functional consequences of aHUS-associated C3 mutations relative to the interaction of C3 with complement regulatory proteins mediating cofactor activity.
© 2015 by The American Society of Hematology.
Figures






Comment in
-
Mutations in complement C3 from aHUS patients.Blood. 2015 Apr 9;125(15):2316-8. doi: 10.1182/blood-2015-02-625285. Blood. 2015. PMID: 25858888
References
-
- Roumenina LT, Loirat C, Dragon-Durey MA, Halbwachs-Mecarelli L, Sautes-Fridman C, Fremeaux-Bacchi V. Alternative complement pathway assessment in patients with atypical HUS. J Immunol Methods. 2011;365(1-2):8–26. - PubMed
-
- Farries TC, Seya T, Harrison RA, Atkinson JP. Competition for binding sites on C3b by CR1, CR2, MCP, factor B and factor H. Complement Inflamm. 1990;7(1):30–41. - PubMed
-
- Oran AE, Isenman DE. Identification of residues within the 727-767 segment of human complement component C3 important for its interaction with factor H and with complement receptor 1 (CR1, CD35). J Biol Chem. 1999;274(8):5120–5130. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- RC2 HL102923/HL/NHLBI NIH HHS/United States
- UC2 HL102926/HL/NHLBI NIH HHS/United States
- UC2 HL103010/HL/NHLBI NIH HHS/United States
- RC2 HL102926/HL/NHLBI NIH HHS/United States
- U54-HL112303/HL/NHLBI NIH HHS/United States
- HL 102924/HL/NHLBI NIH HHS/United States
- G0701325/Medical Research Council/United Kingdom
- RC2 HL102924/HL/NHLBI NIH HHS/United States
- HL-102926/HL/NHLBI NIH HHS/United States
- UC2 HL102923/HL/NHLBI NIH HHS/United States
- R01-AIO41592/PHS HHS/United States
- HL 103010/HL/NHLBI NIH HHS/United States
- UC2 HL102924/HL/NHLBI NIH HHS/United States
- HL-102925/HL/NHLBI NIH HHS/United States
- U54 HL112303/HL/NHLBI NIH HHS/United States
- RC2 HL103010/HL/NHLBI NIH HHS/United States
- HL-102923/HL/NHLBI NIH HHS/United States
- RC2 HL102925/HL/NHLBI NIH HHS/United States
- UC2 HL102925/HL/NHLBI NIH HHS/United States
- T32 GM008629/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
