

Cloud computing approaches for prediction of ligand binding poses and pathways
- PMID: 25608737
- PMCID: PMC4302315
- DOI: 10.1038/srep07918
Cloud computing approaches for prediction of ligand binding poses and pathways
Abstract
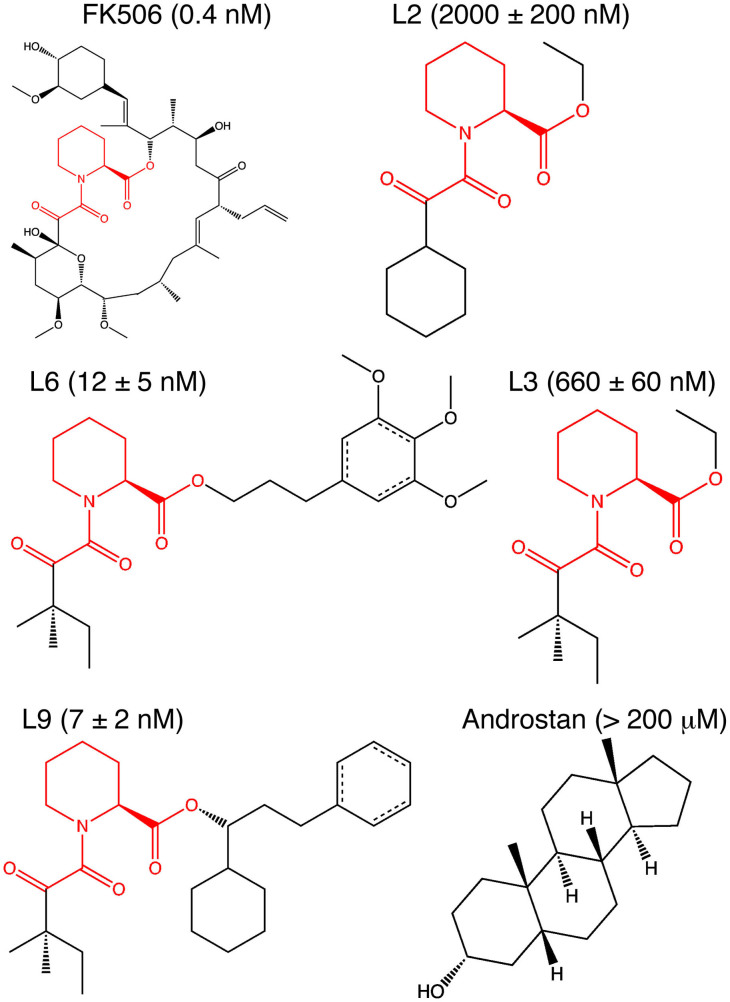
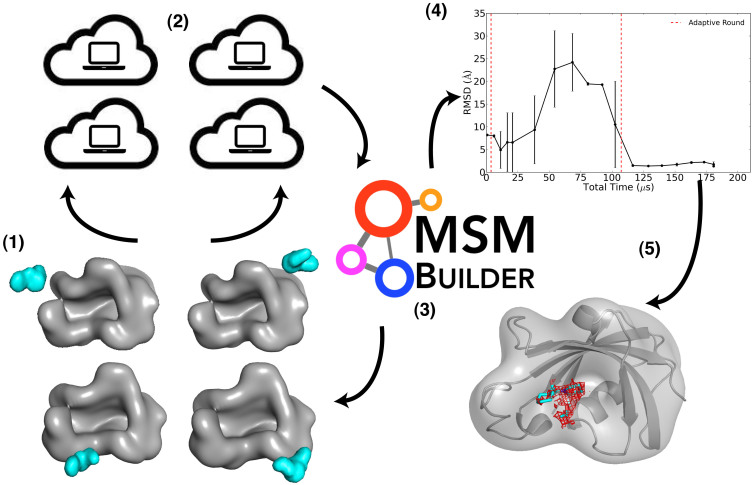
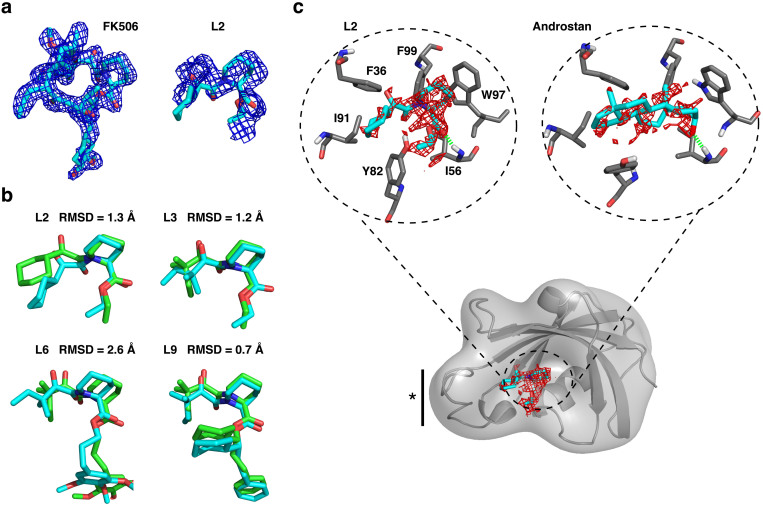
We describe an innovative protocol for ab initio prediction of ligand crystallographic binding poses and highly effective analysis of large datasets generated for protein-ligand dynamics. We include a procedure for setup and performance of distributed molecular dynamics simulations on cloud computing architectures, a model for efficient analysis of simulation data, and a metric for evaluation of model convergence. We give accurate binding pose predictions for five ligands ranging in affinity from 7 nM to > 200 μM for the immunophilin protein FKBP12, for expedited results in cases where experimental structures are difficult to produce. Our approach goes beyond single, low energy ligand poses to give quantitative kinetic information that can inform protein engineering and ligand design.
Figures
References
-
- Hoeppner A., Schmitt L. & Smits S. Proteins and their ligands: Their importance and how to crystallize them. Advanced Topics on Crystal Growth Ferreira, S. O. (ed.) (InTech, 2013).
-
- Schneider G. Virtual screening: an endless staircase? Nat. Rev. Drug Discovery 9, 273–276 (2010). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
