

HDAC2 selectively regulates FOXO3a-mediated gene transcription during oxidative stress-induced neuronal cell death
- PMID: 25609639
- PMCID: PMC6605534
- DOI: 10.1523/JNEUROSCI.2444-14.2015
HDAC2 selectively regulates FOXO3a-mediated gene transcription during oxidative stress-induced neuronal cell death
Abstract
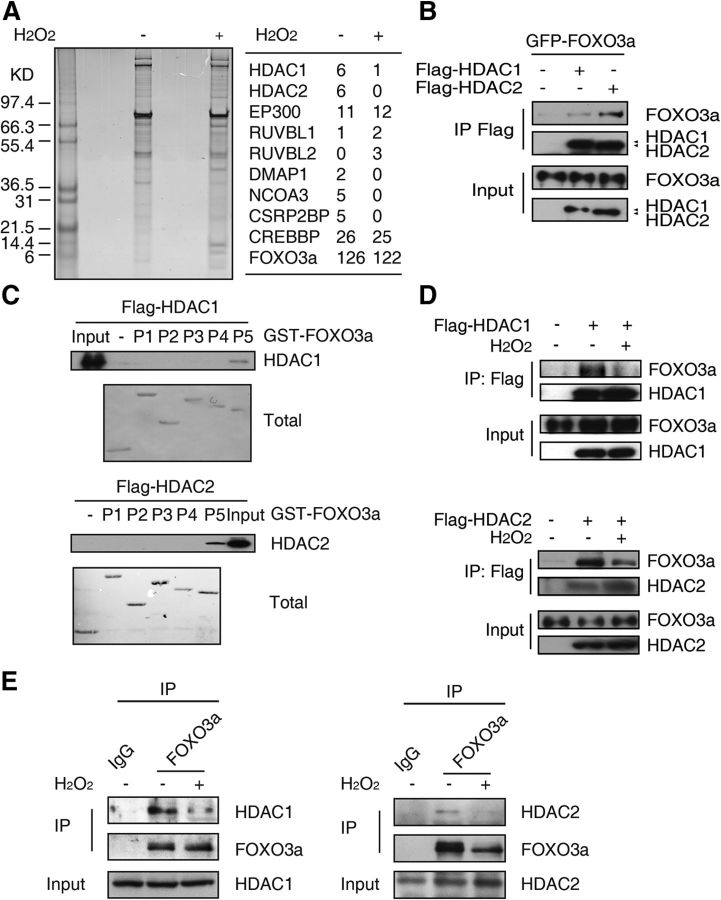
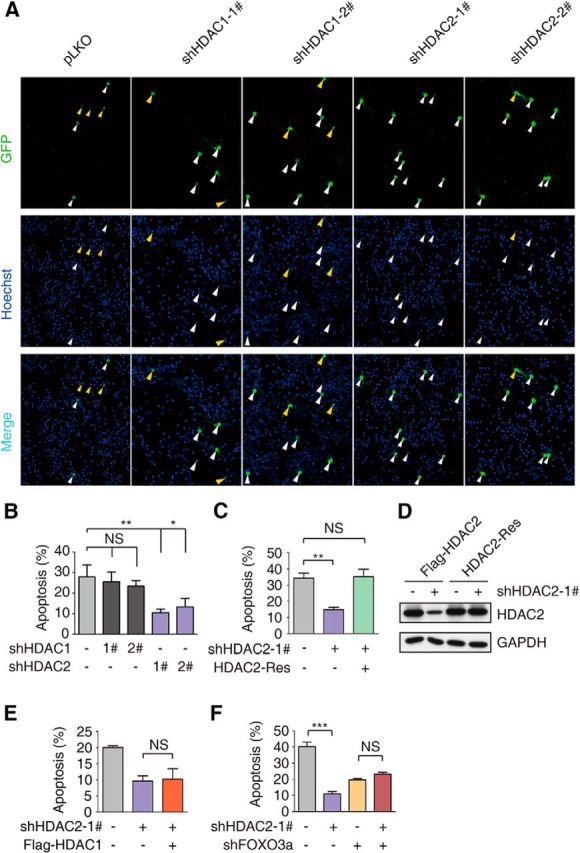
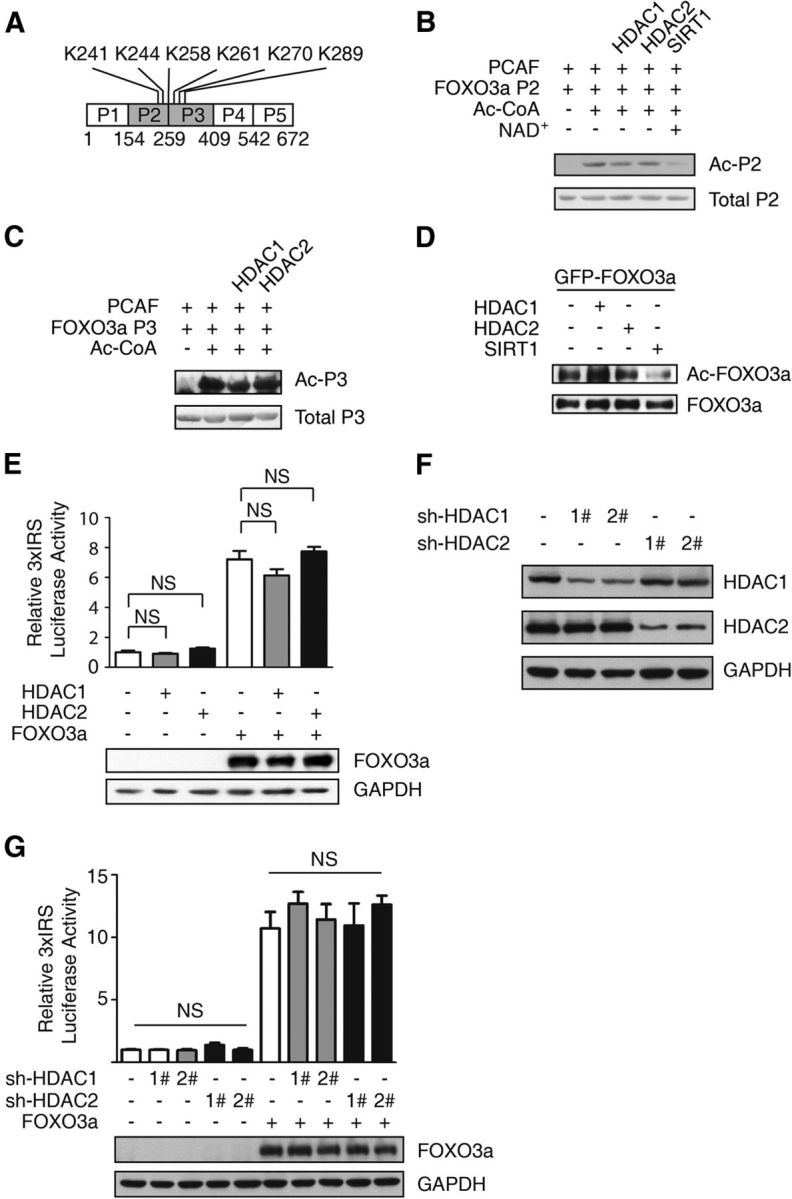
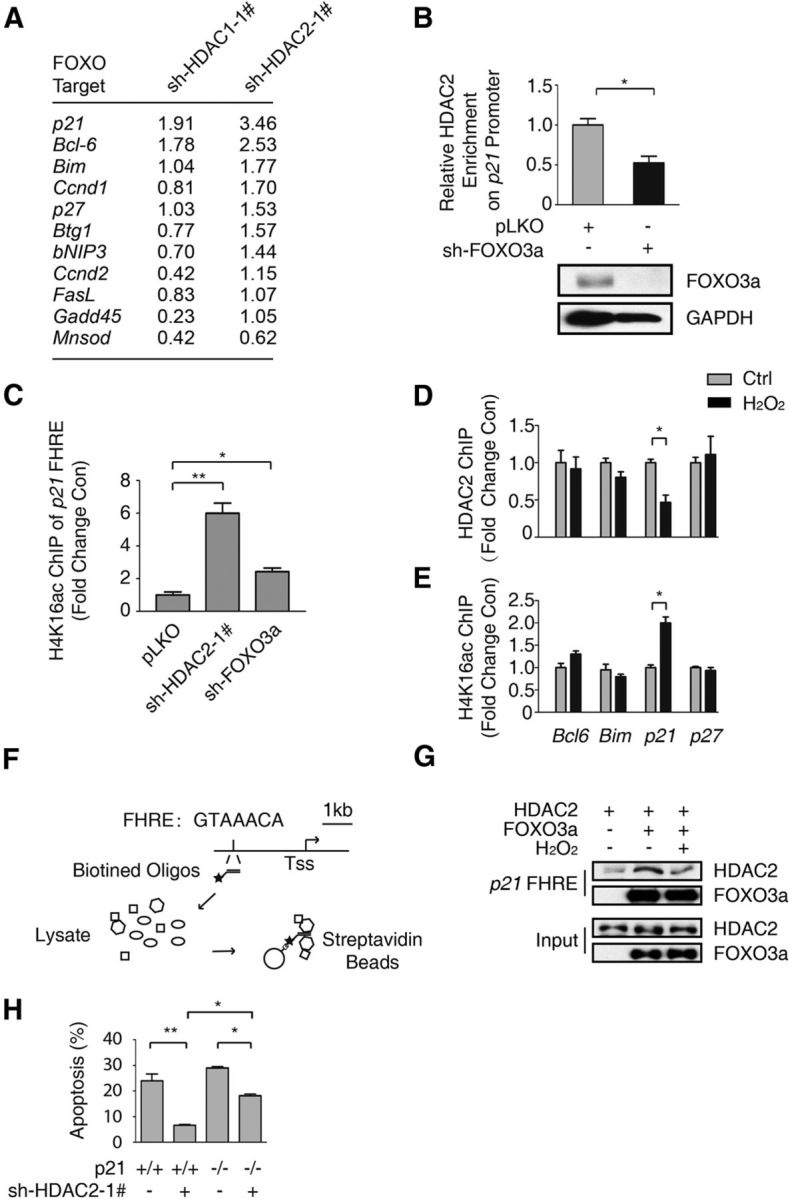
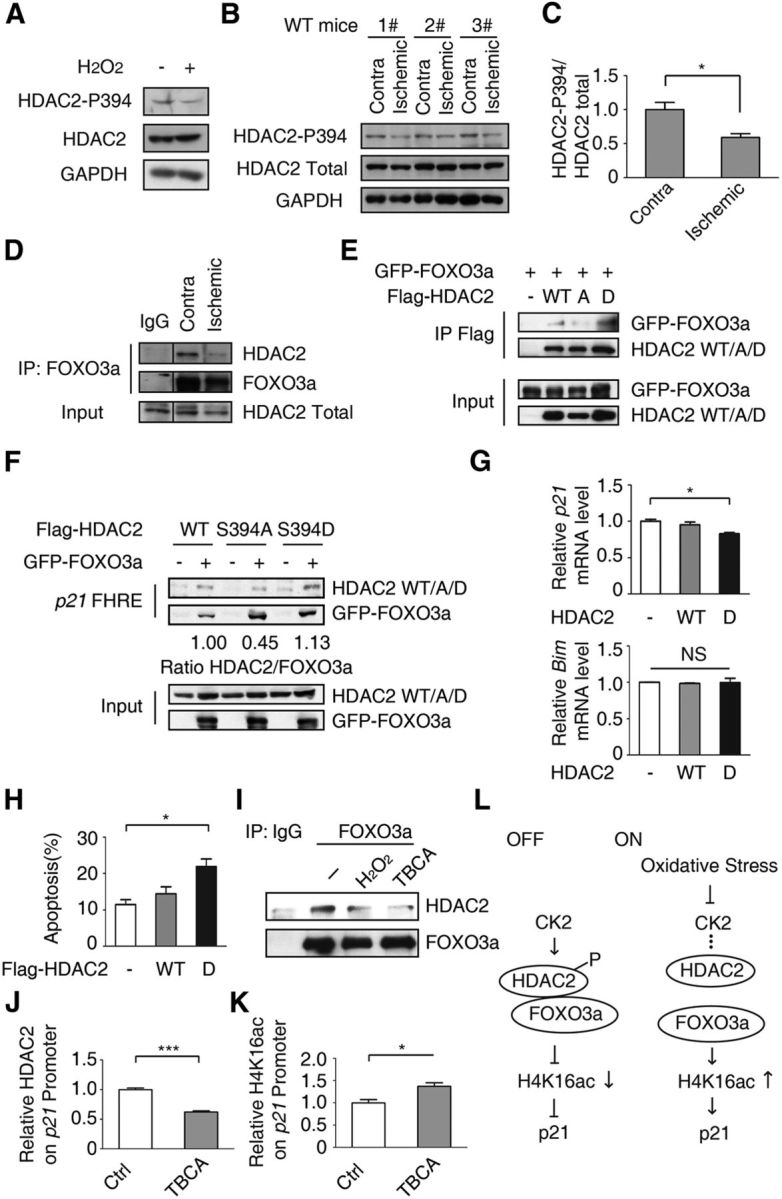
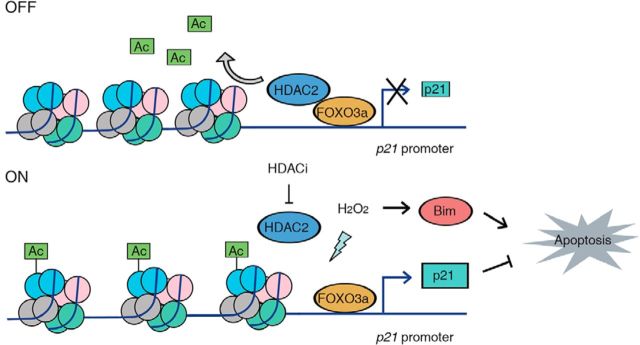
All neurodegenerative diseases are associated with oxidative stress-induced neuronal death. Forkhead box O3a (FOXO3a) is a key transcription factor involved in neuronal apoptosis. However, how FOXO3a forms complexes and functions in oxidative stress processing remains largely unknown. In the present study, we show that histone deacetylase 2 (HDAC2) forms a physical complex with FOXO3a, which plays an important role in FOXO3a-dependent gene transcription and oxidative stress-induced mouse cerebellar granule neuron (CGN) apoptosis. Interestingly, we also found that HDAC2 became selectively enriched in the promoter region of the p21 gene, but not those of other target genes, and inhibited FOXO3a-mediated p21 transcription. Furthermore, we found that oxidative stress reduced the interaction between FOXO3a and HDAC2, leading to an increased histone H4K16 acetylation level in the p21 promoter region and upregulated p21 expression in a manner independent of p53 or E2F1. Phosphorylation of HDAC2 at Ser 394 is important for the HDAC2-FOXO3a interaction, and we found that cerebral ischemia/reperfusion reduced phosphorylation of HDAC2 at Ser 394 and mitigated the HDAC2-FOXO3a interaction in mouse brain tissue. Our study reveals the novel regulation of FOXO3a-mediated selective gene transcription via epigenetic modification in the process of oxidative stress-induced cell death, which could be exploited therapeutically.
Keywords: FOXO3a; HDAC2; oxidative stress; p21; transcription.
Copyright © 2015 the authors 0270-6474/15/351250-10$15.00/0.
Figures

References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous