

Delineating community outbreaks of Salmonella enterica serovar Typhimurium by use of whole-genome sequencing: insights into genomic variability within an outbreak
- PMID: 25609719
- PMCID: PMC4365247
- DOI: 10.1128/JCM.03235-14
Delineating community outbreaks of Salmonella enterica serovar Typhimurium by use of whole-genome sequencing: insights into genomic variability within an outbreak
Abstract
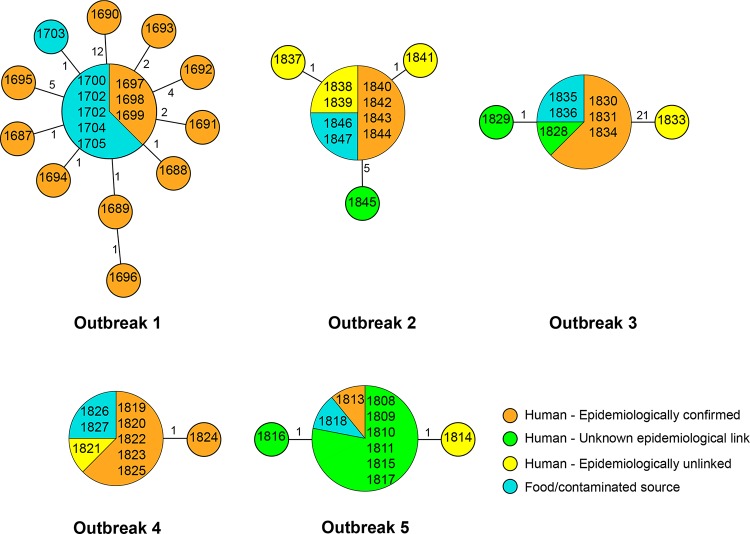
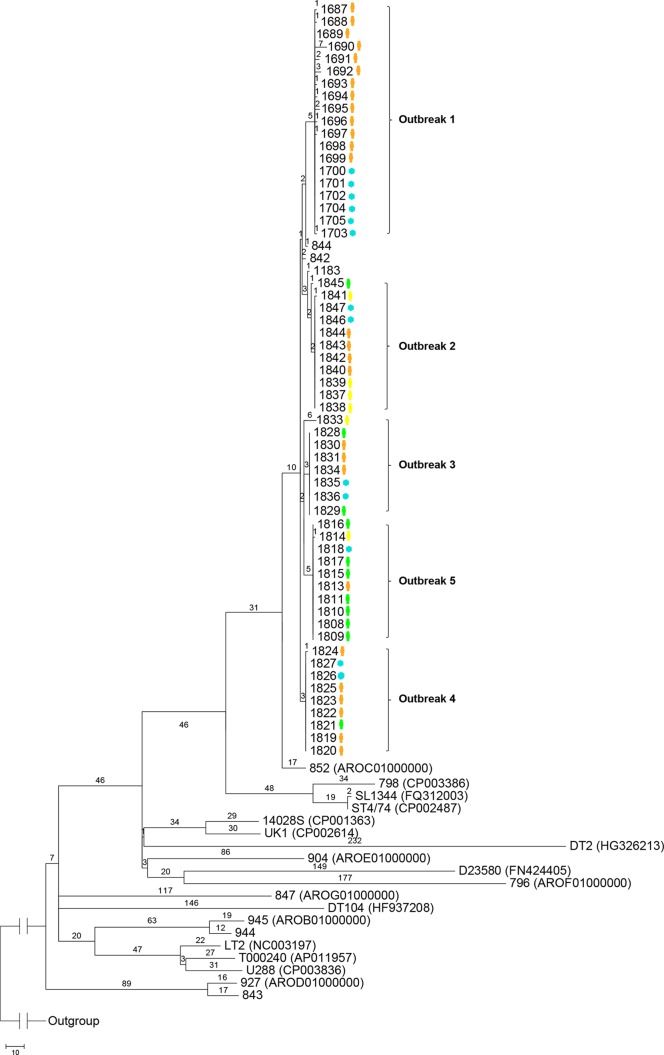
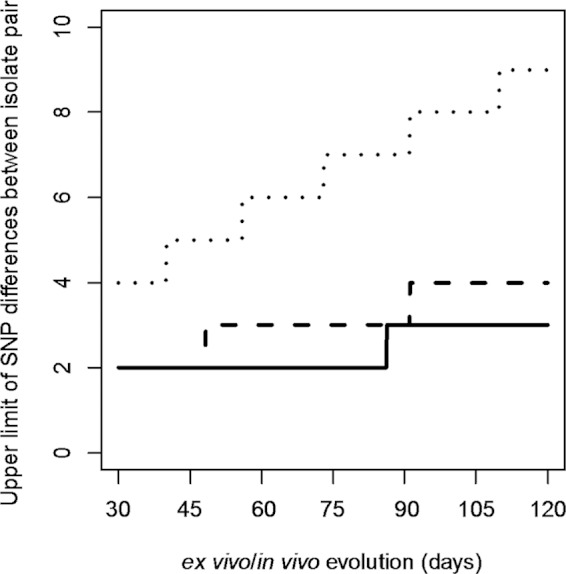
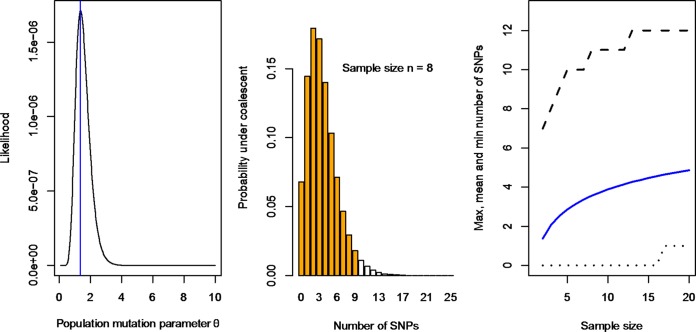
Whole-genome next-generation sequencing (NGS) was used to retrospectively examine 57 isolates from five epidemiologically confirmed community outbreaks (numbered 1 to 5) caused by Salmonella enterica serovar Typhimurium phage type DT170. Most of the human and environmental isolates confirmed epidemiologically to be involved in the outbreaks were either genomically identical or differed by one or two single nucleotide polymorphisms (SNPs), with the exception of those in outbreak 1. The isolates from outbreak 1 differed by up to 12 SNPs, which suggests that the food source of the outbreak was contaminated with more than one strain while each of the other four outbreaks was caused by a single strain. In addition, NGS analysis ruled in isolates that were initially not considered to be linked with the outbreak, which increased the total outbreak size by 107%. The mutation process was modeled by using known mutation rates to derive a cutoff value for the number of SNP difference to determine whether or not a case was part of an outbreak. For an outbreak with less than 1 month of ex vivo/in vivo evolution time, the maximum number of SNP differences between isolates is two or four using the lowest or highest mutation rate, respectively. NGS of S. Typhimurium significantly increases the resolution of investigations of community outbreaks. It can also inform a more targeted public health response by providing important supplementary evidence that cases of disease are or are not associated with food-borne outbreaks of S. Typhimurium.
Copyright © 2015, American Society for Microbiology. All Rights Reserved.
Figures
Comment in
-
Integration of whole-genome sequencing into infection control practices: the potential and the hurdles.J Clin Microbiol. 2015 Apr;53(4):1054-5. doi: 10.1128/JCM.00349-15. Epub 2015 Feb 11. J Clin Microbiol. 2015. PMID: 25673795 Free PMC article.
References
-
- Sintchenko V, Wang Q, Howard P, Ha CW, Kardamanidis K, Musto J, Gilbert GL. 2012. Improving resolution of public health surveillance for human Salmonella enterica serovar Typhimurium infection: 3 years of prospective multiple-locus variable-number tandem-repeat analysis (MLVA). BMC Infect Dis 12:78. doi:10.1186/1471-2334-12-78. - DOI - PMC - PubMed
-
- Larsson J, Torpdahl M, Petersen R, Sørensen G, Lindsted B, Nielsen E. 2009. Development of a new nomenclature for Salmonella Typhimurium multilocus variable number of tandem repeats analysis (MLVA). Eurosurveillance 14:pii=19174 http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=19174. - PubMed
-
- Bryant JM, Schurch AC, van Deutekom H, Harris SR, de Beer JL, de Jager V, Kremer K, van Hijum SA, Siezen RJ, Borgdorff M, Bentley SD, Parkhill J, van Soolingen D. 2013. Inferring patient to patient transmission of Mycobacterium tuberculosis from whole genome sequencing data. BMC Infect Dis 13:110. doi:10.1186/1471-2334-13-110. - DOI - PMC - PubMed
-
- Eyre DW, Golubchik T, Gordon NC, Bowden R, Piazza P, Batty EM, Ip CL, Wilson DJ, Didelot X, O'Connor L, Lay R, Buck D, Kearns AM, Shaw A, Paul J, Wilcox MH, Donnelly PJ, Peto TE, Walker AS, Crook DW. 2012. A pilot study of rapid benchtop sequencing of Staphylococcus aureus and Clostridium difficile for outbreak detection and surveillance. BMJ Open 2:pii=e001124. doi:10.1136/bmjopen-2012-001124. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
