

Novel mutations expand the clinical spectrum of DYNC1H1-associated spinal muscular atrophy
- PMID: 25609763
- PMCID: PMC4336105
- DOI: 10.1212/WNL.0000000000001269
Novel mutations expand the clinical spectrum of DYNC1H1-associated spinal muscular atrophy
Abstract
Objective: To expand the clinical phenotype of autosomal dominant congenital spinal muscular atrophy with lower extremity predominance (SMA-LED) due to mutations in the dynein, cytoplasmic 1, heavy chain 1 (DYNC1H1) gene.
Methods: Patients with a phenotype suggestive of a motor, non-length-dependent neuronopathy predominantly affecting the lower limbs were identified at participating neuromuscular centers and referred for targeted sequencing of DYNC1H1.
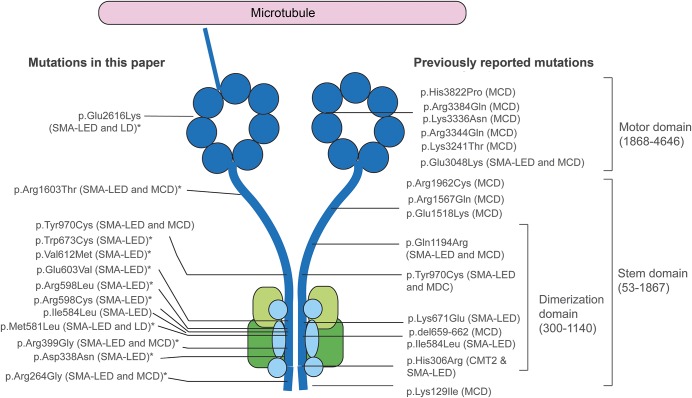
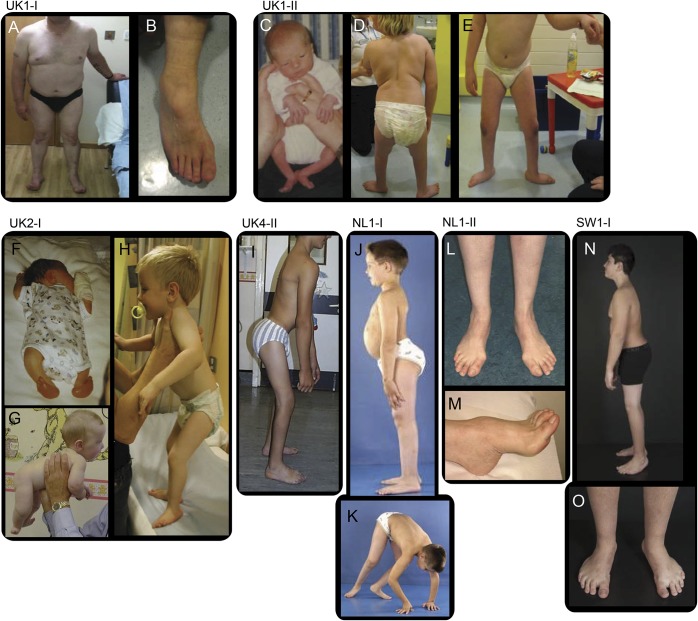
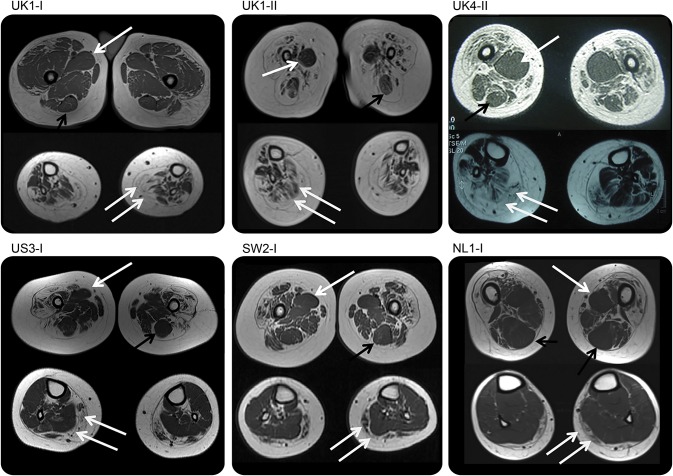
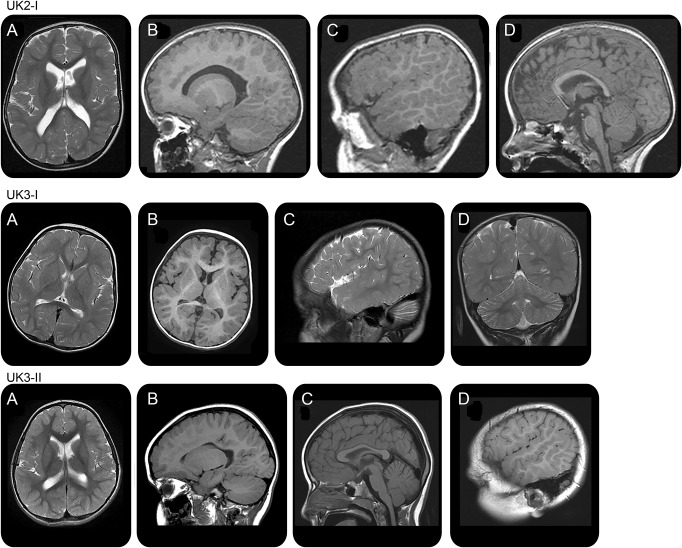
Results: We report a cohort of 30 cases of SMA-LED from 16 families, carrying mutations in the tail and motor domains of DYNC1H1, including 10 novel mutations. These patients are characterized by congenital or childhood-onset lower limb wasting and weakness frequently associated with cognitive impairment. The clinical severity is variable, ranging from generalized arthrogryposis and inability to ambulate to exclusive and mild lower limb weakness. In many individuals with cognitive impairment (9/30 had cognitive impairment) who underwent brain MRI, there was an underlying structural malformation resulting in polymicrogyric appearance. The lower limb muscle MRI shows a distinctive pattern suggestive of denervation characterized by sparing and relative hypertrophy of the adductor longus and semitendinosus muscles at the thigh level, and diffuse involvement with relative sparing of the anterior-medial muscles at the calf level. Proximal muscle histopathology did not always show classic neurogenic features.
Conclusion: Our report expands the clinical spectrum of DYNC1H1-related SMA-LED to include generalized arthrogryposis. In addition, we report that the neurogenic peripheral pathology and the CNS neuronal migration defects are often associated, reinforcing the importance of DYNC1H1 in both central and peripheral neuronal functions.
© 2015 American Academy of Neurology.
Figures
References
-
- Frijns CJ, Van Deutekom J, Frants RR, Jennekens FG. Dominant congenital benign spinal muscular atrophy. Muscle Nerve 1994;17:192–197. - PubMed
-
- Van der Vleuten AJ, van Ravenswaaij-Arts CM, Frijns CJ, et al. Localisation of the gene for a dominant congenital spinal muscular atrophy predominantly affecting the lower limbs to chromosome 12q23-q24. Eur J Hum Genet 1998;6:376–382. - PubMed
-
- Mercuri E, Messina S, Kinali M, et al. Congenital form of spinal muscular atrophy predominantly affecting the lower limbs: a clinical and muscle MRI study. Neuromuscul Disord 2004;14:125–129. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical