

Modulation of a pore in the capsid of JC polyomavirus reduces infectivity and prevents exposure of the minor capsid proteins
- PMID: 25609820
- PMCID: PMC4403419
- DOI: 10.1128/JVI.00089-15
Modulation of a pore in the capsid of JC polyomavirus reduces infectivity and prevents exposure of the minor capsid proteins
Abstract
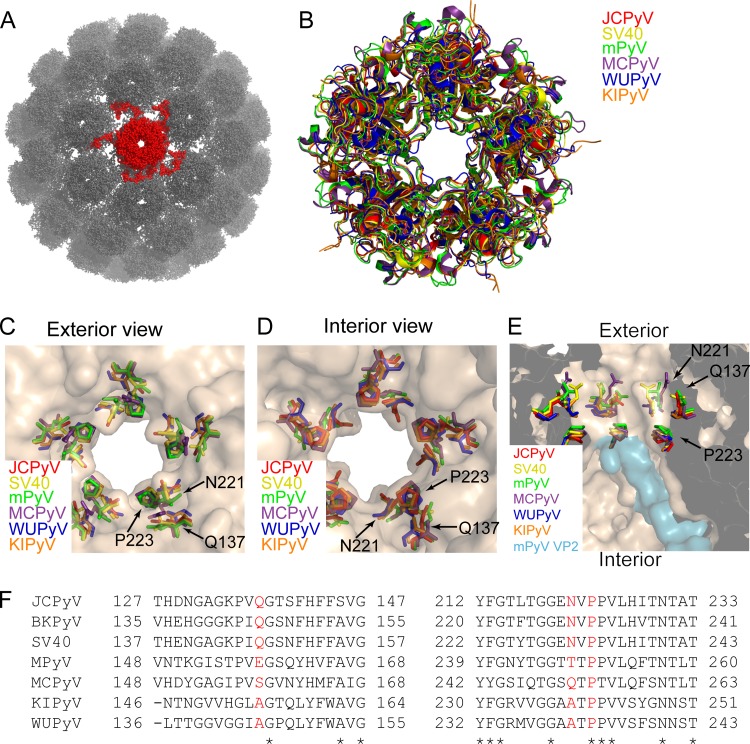
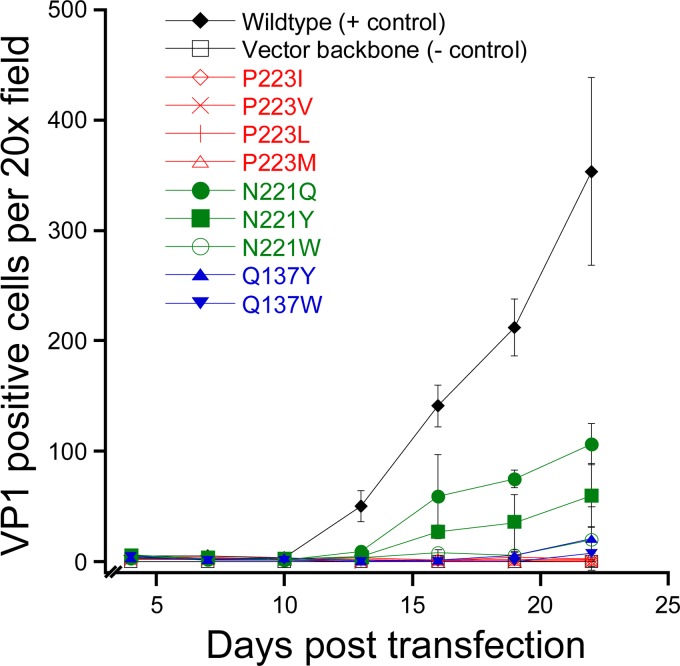
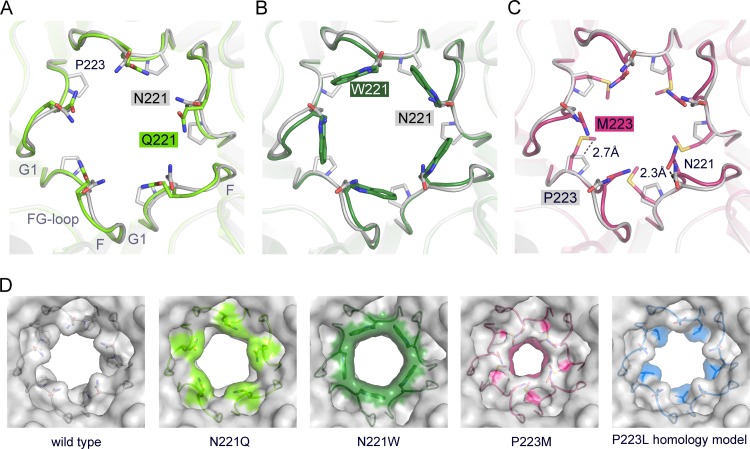
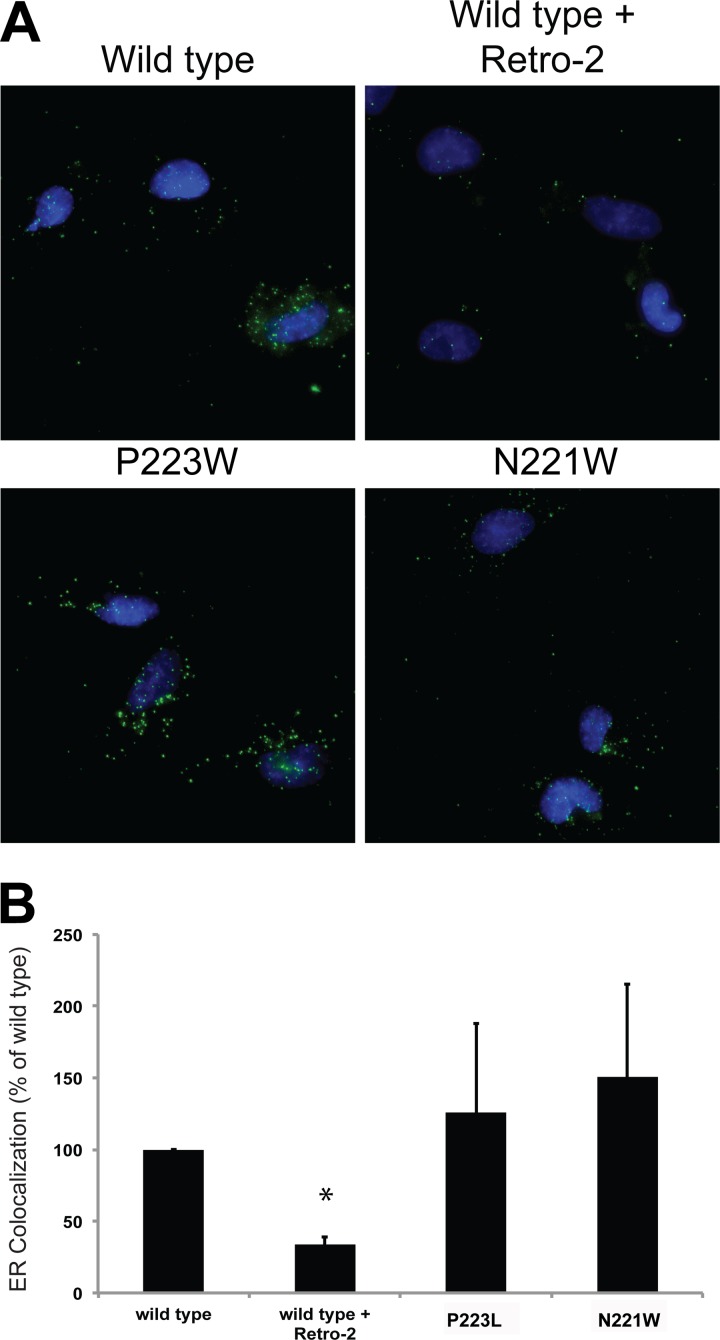
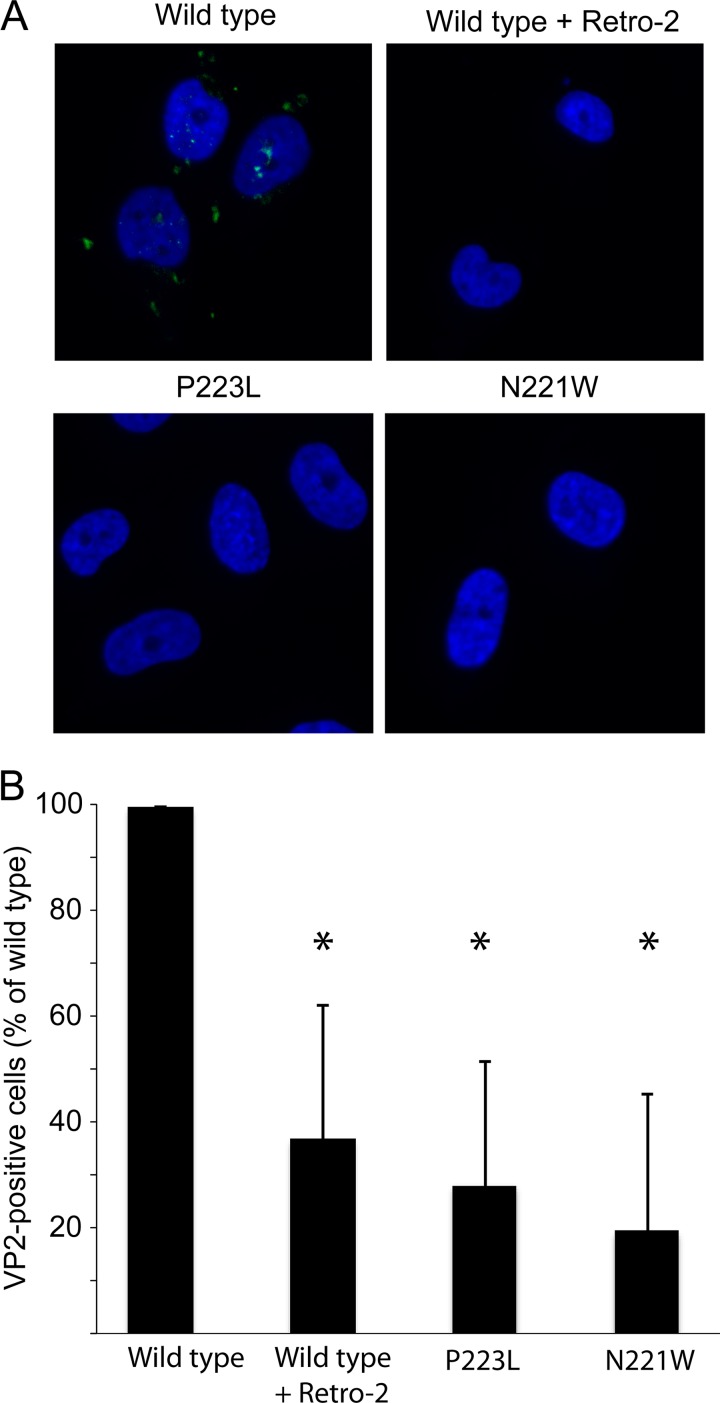
JC polyomavirus (JCPyV) infection of immunocompromised individuals results in the fatal demyelinating disease progressive multifocal leukoencephalopathy (PML). The viral capsid of JCPyV is composed primarily of the major capsid protein virus protein 1 (VP1), and pentameric arrangement of VP1 monomers results in the formation of a pore at the 5-fold axis of symmetry. While the presence of this pore is conserved among polyomaviruses, its functional role in infection or assembly is unknown. Here, we investigate the role of the 5-fold pore in assembly and infection of JCPyV by generating a panel of mutant viruses containing amino acid substitutions of the residues lining this pore. Multicycle growth assays demonstrated that the fitness of all mutants was reduced compared to that of the wild-type virus. Bacterial expression of VP1 pentamers containing substitutions to residues lining the 5-fold pore did not affect pentamer assembly or prevent association with the VP2 minor capsid protein. The X-ray crystal structures of selected pore mutants contained subtle changes to the 5-fold pore, and no other changes to VP1 were observed. Pore mutant pseudoviruses were not deficient in assembly, packaging of the minor capsid proteins, or binding to cells or in transport to the host cell endoplasmic reticulum. Instead, these mutant viruses were unable to expose VP2 upon arrival to the endoplasmic reticulum, a step that is critical for infection. This study demonstrated that the 5-fold pore is an important structural feature of JCPyV and that minor modifications to this structure have significant impacts on infectious entry.
Importance: JCPyV is an important human pathogen that causes a severe neurological disease in immunocompromised individuals. While the high-resolution X-ray structure of the major capsid protein of JCPyV has been solved, the importance of a major structural feature of the capsid, the 5-fold pore, remains poorly understood. This pore is conserved across polyomaviruses and suggests either that these viruses have limited structural plasticity in this region or that this pore is important in infection or assembly. Using a structure-guided mutational approach, we showed that modulation of this pore severely inhibits JCPyV infection. These mutants do not appear deficient in assembly or early steps in infectious entry and are instead reduced in their ability to expose a minor capsid protein in the host cell endoplasmic reticulum. Our work demonstrates that the 5-fold pore is an important structural feature for JCPyV.
Copyright © 2015, American Society for Microbiology. All Rights Reserved.
Figures

References
-
- Ferenczy MW, Marshall LJ, Nelson CD, Atwood WJ, Nath A, Khalili K, Major EO. 2012. Molecular biology, epidemiology, and pathogenesis of progressive multifocal leukoencephalopathy, the JC virus-induced demyelinating disease of the human brain. Clin Microbiol Rev 25:471–506. doi:10.1128/CMR.05031-11. - DOI - PMC - PubMed
-
- Zurhein G, Chou SM. 1965. Particles resembling papova viruses in human cerebral demyelinating disease. Science 148:1477–1479. - PubMed
-
- Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed). 2007. Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA.
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
