

Pathologic Regulation of Collagen I by an Aberrant Protein Phosphatase 2A/Histone Deacetylase C4/MicroRNA-29 Signal Axis in Idiopathic Pulmonary Fibrosis Fibroblasts
- PMID: 25612003
- PMCID: PMC4566061
- DOI: 10.1165/rcmb.2014-0150OC
Pathologic Regulation of Collagen I by an Aberrant Protein Phosphatase 2A/Histone Deacetylase C4/MicroRNA-29 Signal Axis in Idiopathic Pulmonary Fibrosis Fibroblasts
Abstract
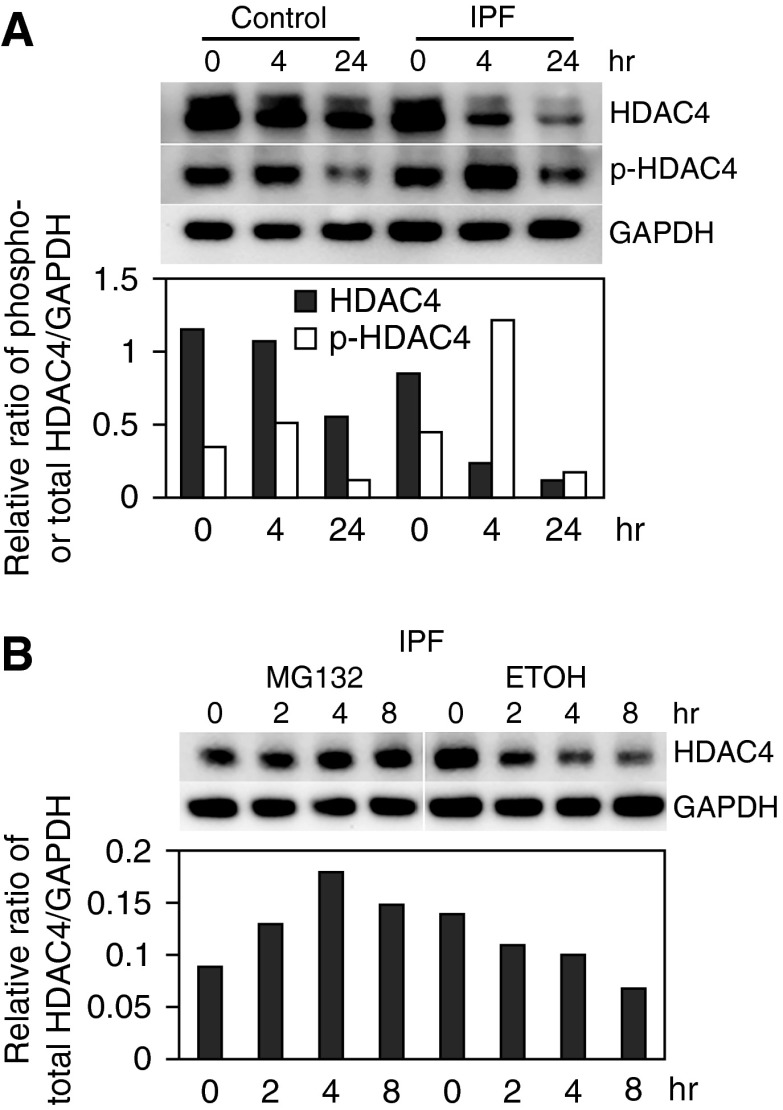
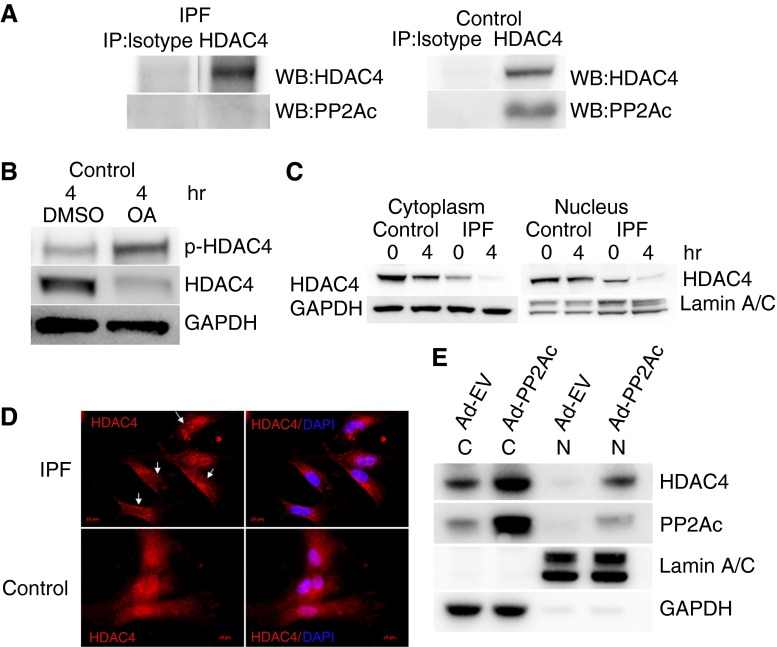
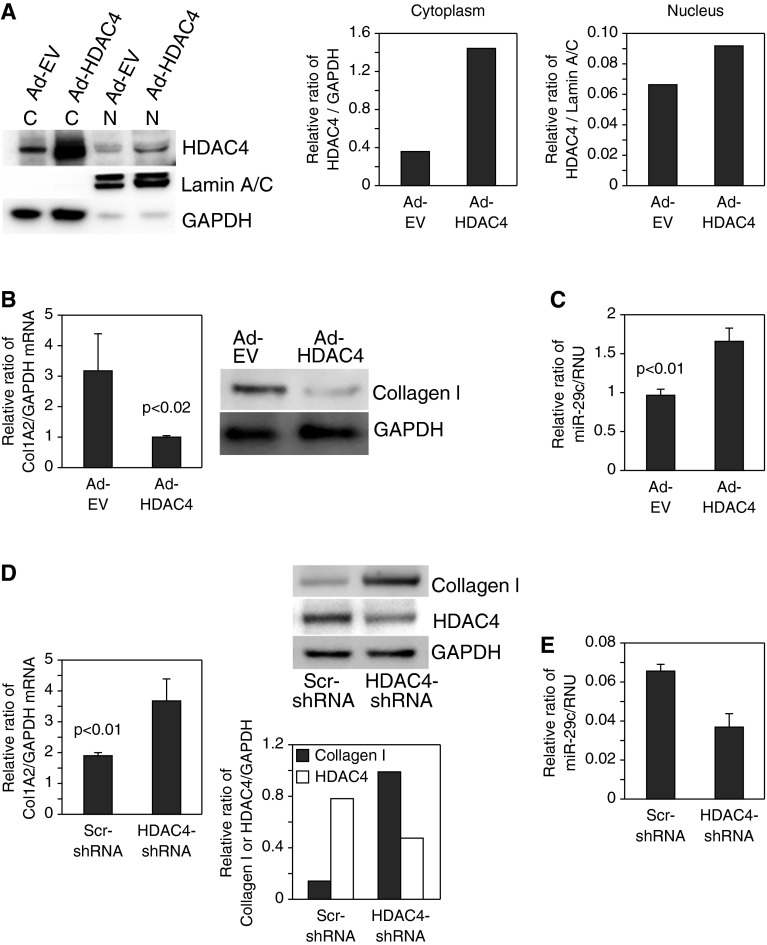
Idiopathic pulmonary fibrosis (IPF) is characterized by the relentless expansion of fibroblasts depositing type I collagen within the alveolar wall and obliterating the alveolar airspace. MicroRNA (miR)-29 is a potent regulator of collagen expression. In IPF, miR-29 levels are low, whereas type I collagen expression is high. However, the mechanism for suppression of miR-29 and increased type I collagen expression in IPF remains unclear. Here we show that when IPF fibroblasts are seeded on polymerized type I collagen, miR-29c levels are suppressed and type I collagen expression is high. In contrast, miR-29c is high and type I collagen expression is low in control fibroblasts. We demonstrate that the mechanism for suppression of miR-29 during IPF fibroblast interaction with polymerized collagen involves inappropriately low protein phosphatase (PP) 2A function, leading to histone deacetylase (HDA) C4 phosphorylation and decreased nuclear translocation of HDAC4. We demonstrate that overexpression of HDAC4 in IPF fibroblasts restored miR-29c levels and decreased type I collagen expression, whereas knocking down HDAC4 in control fibroblasts suppressed miR-29c levels and increased type I collagen expression. Our data indicate that IPF fibroblast interaction with polymerized type I collagen results in an aberrant PP2A/HDAC4 axis, which suppresses miR-29, causing a pathologic increase in type I collagen expression.
Keywords: HDAC4; IPF fibroblasts; PP2A; miR-29; type I collagen.
Figures



References
-
- Coultas DB, Zumwalt RE, Black WC, Sobonya RE. The epidemiology of interstitial lung diseases. Am J Respir Crit Care Med. 1994;150:967–972. - PubMed
-
- Mannino DM, Etzel RA, Parrish RG. Pulmonary fibrosis deaths in the United States, 1979–1991: an analysis of multiple-cause mortality data. Am J Respir Crit Care Med. 1996;153:1548–1552. - PubMed
-
- Noble PW. Idiopathic pulmonary fibrosis: natural history and prognosis. Clin Chest Med. 2006;27(1) Suppl 1:S11–S16. - PubMed
-
- Raghu G, Weycker D, Edelsberg J, Bradford WZ, Oster G. Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2006;174:810–816. - PubMed
-
- King TE, Costabel U, Cordier JF, DoPico GA, DuBois RM, Lynch D, Lynch JP, III, Myers J, Panos R, Raghu G, et al. American Thoracic Society. Idiopathic pulmonary fibrosis: diagnosis and treatment: International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS) Am J Respir Crit Care Med. 2000;161:646–664. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
